



Достаточно хорошо известно, что халькогениды металлов представляют значительный интерес в различных технологических областях, таких как микроэлектроника, электрооптика, нелинейная оптика, фотокатализ, фотоэлектрохимия. В частности, сульфид свинца вследствие почти идеальной монокристаллической формы – один из самых используемых и исследуемых полупроводников. Благодаря наличию прямых электронных переходов, PbS нашел применение как инфракрасный детектор, используется в лазерных технологиях [6, 1], а также как материал для исследования свойств и поиска прикладного применения квантовых точек [2].
Наряду с экспериментальными исследованиями развивается теория фононных спектров с использованием моделей – жестких ионов, деформируемых диполей, зарядов на связях, а также в рамках теории функционала плотности [3, 4].
Цель работы – анализ применимости теории функционала плотности к оценке геометрических, спектральных и термодинамических характеристик халькогенидов свинца.
Материалы и методы исследования
При проведении квантово-химических расчетов нами использован стандартный пакет программ GAUSSIAN’03W [7]. Расчеты проводили гибридным методом функционала плотности B3LYP, с обменным функционалом Беке В3 и корреляционным функционалом Ли, Янга и Пара (LYP) [8, 9]. Этот метод является общепринятым в настоящее время для описания термодинамических характеристик и колебательных спектров [10]. В качестве базисного состояния использован псевдопотенциал SDD для атома свинца и D95 double-dzeta базис для остальных атомов. Геометрии всех рассчитанных молекул были полностью оптимизированы, отсутствие мнимых частот колебаний подтверждало их стационарный характер. Энергии диссоциации рассчитанных соединений были скорректированы с учетом нулевой колебательной энергии (ZPVE) и приведены к стандартным условиям (298, 15К, 1 атм) с использованием термической поправки к энтальпии и свободной энергии. Нами также были проведены расчеты по программе ADF'2004 (Амстердамский функционал плотности) [10]. Мы использовали обменный функционал OPTX, объединенный с корреляционным функционалом PBE, используя полноэлектронный трипл-ζ с учетом поляризации базисного набора Слейтеровских орбиталей (BP86/TZ2P+) [12]. Все вышеприведенные расчеты проведены для соединений свинца, серы, селена и теллура, а также PbS, PbSе и PbTe, находящихся в газовой фазе. Расчет электронной структуры и характеристик PbS, PbSе и PbTe, находящихся в кристаллическом состоянии проведен полно-потенциальным линейным методом присоединенных плоских волн (FLAPW, код WIEN2k [13]) с обобщенной градиентной аппроксимацией (GGA) обменно-корреляционного потенциала [14] с полной структурной оптимизацией. Плотности электронных состояний получены методом тетраэдров [15].
В этом случае для расчетов использовались кубические (типа В1, простр. группа № 225, Fm-3m), моносульфид, моноселенид и монотеллурид свинца PbS, PbSe, PbTe; атомные координаты: Pb (0;0;0) и S, Se,Te (1/2;1/2;1/2).
Результаты исследования и их обсуждение
Для оценки «качества» квантово-химических расчетов халькогенидов свинца необходимо опираться на некоторые физико-химические свойства подобных соединений, для которых известны экспериментальные достаточно надежные значения. В качестве последних нами использованы длины связей, частоты валентных и деформационных колебаний в ИК-спектрах соединений свинца, серы, селена и теллура, а также экспериментальные значения ширины запрещенной зоны кристаллических халькогенидов и энергии диссоциации соединений серы, селена и теллура в газовой фазе (табл. 1). Из результатов расчета следует, что оптимизированные методом B3LYP/SDD длины связей в соединениях серы, селена и теллура пропорциональны экспериментальным значениям [5], и наблюдаются хорошие корреляционные соотношения между ними:
Таблица 1
Экспериментальные и рассчитанные энергии диссоциации соединений серы, селена и теллура
|
Молекула |
Экспериментальная энергия диссоциации связи, кДж/моль |
Рассчитанная методом ADF энергия диссоциации связи, кДж/моль |
Рассчитанная методом B3LYP/6-31G(d) энергия диссоциации связи, кДж/моль |
|
1 |
2 |
3 |
4 |
|
SF |
343 |
572 |
527 |
|
SCl |
277 |
401 |
432 |
|
SH |
345 |
495 |
543 |
|
SO |
522 |
781 |
799 |
|
SP |
444 |
718 |
743 |
|
SC |
714 |
994 |
996 |
|
TeB |
354 |
485 |
477 |
|
SeAl |
338 |
468 |
438 |
|
TeAl |
268 |
343 |
372 |
|
SeIn |
247 |
359 |
414 |
|
TeIn |
218 |
326 |
364 |
|
SeP |
364 |
505 |
574 |
|
SeAs |
352 |
490 |
564 |
|
TeAs |
312 |
445 |
519 |
|
SeS |
371 |
547 |
490 |
|
TeS |
339 |
502 |
469 |
|
SeTe |
292 |
468 |
446 |
|
SeO |
465 |
694 |
623 |
|
SeC |
590 |
869 |
802 |
|
Se2 |
333 |
510 |
468 |
|
SeH |
311 |
493 |
439 |
|
SeSi |
534 |
731 |
694 |
|
SeGe |
485 |
694 |
673 |
|
SeSn |
401 |
635 |
631 |
|
Te2 |
260 |
390 |
422 |
|
TeSi |
448 |
631 |
623 |
|
TeO |
376 |
550 |
602 |
|
TeCu |
229 |
320 |
351 |
|
TeGe |
430 |
610 |
614 |
Для соединений серы R(эксп.) = 0.06+0.994R (расч.) r = 0.999; s = 0.01; n = 6 (1)
Для соединений селена R(эксп.) = 0.957R (расч.) r = 0.995; s = 0.04; n = 18 (2)
Для соединений теллура R(эксп.) = 0.969R (расч.) r = 0.993; s = 0.04; n = 10 (3)
Здесь и далее r – коэффициент корреляции, s – стандартное отклонение, и n – число соединений.
Для галогенидов свинца из результатов расчета следует, что оптимизированные методом B3LYP/SDD валентные углы Hal-Pb-Hal близки к экспериментальным значениям [5] (около 98-100 о), а длины связей Pb-Hal (Hal = F, Cl, Br, I, Me) Pb(II) и Pb(IV) соединений дают хорошее корреляционное соотношение (рис. 1).
Рассчитанные обоими методами частоты ИК- и Рамановских спектров галогенидов свинца достаточно близки к экспериментальным значениям (рис. 2, 3) с коэффициентом корреляции 0.998 и стандартным отклонением 9.
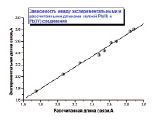
Рис. 1. Зависимость между экспериментальными и рассчитанными методом B3LYP/SDD длинами связей соединений Pb(II) и Pb(IV)
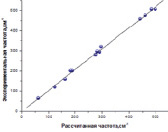
Рис. 2. Зависимость между экспериментальными и рассчитанными методом B3LYP/SDD ИК частотами галогенидов свинца
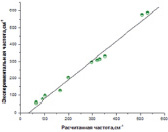
Рис. 3. Зависимость между экспериментальными и рассчитанными методом ADF ИК частотами галогенидов свинца
Кроме того, рассчитанные частоты ИК- и Рамановских спектров соединений селена и теллура хорошо ложатся на прямолинейные зависимости [16]:
Для соединений серы ω(эксп.) = 46+0.960 ω(расч.) r = 0.998; s = 35; n = 34 (4)
Для соединений селена ω(эксп.) = 59+0.996 ω(расч.) r = 0.993; s = 57; n = 91 (5)
Для соединений теллура ω(эксп.) = 21+1.07 ω(расч.) r = 0.993; s = 27; n = 67 (6)
Необходимо отметить, что близкий к единице в соотношениях (1-6) коэффициент перед рассчитанными параметрами указывает на высокую надежность результатов использования функционала плотности и базисного SDD состояния.
Нами получены также достаточно надежные линейные корреляционные зависимости (7 и 8), которые свидетельствует о том, что проведенные расчеты соединений селена и теллура методами B3LYP/SDD и ВР86/TZ2P+ правильно описывают не только геометрию, но и термодинамические параметры, в частности, энергии диссоциации (D0). Энергия диссоциации соединений серы, селена и теллура определялась как разница между энергией соединения и энергиями составляющих их атомов с поправкой на нулевую энергию. Рассчитанные энергии диссоциации, отнесенные к числу связей, сравнивались с экспериментальными средними энергиями связей элемент-сера, селен и теллур [17] (Табл. 1). В результате получены корреляционные соотношения на основании обоих методов расчета в программах GAUSSIAN и ADF (рис. 3, 4):
GAUSSIAN: D0 (эксп.) = -25 +0.73 D0 (расч.) r = 0.963; s = 31; n = 29 (7)
ADF: D0 (эксп.) = 1.8 +0.68 D0 (расч.) r = 0.980; s = 23; n = 29 (8)
Полученные корреляционные соотношения указывают на то, что рассчитанные разными методами термодинамические параметры соединений серы, селена и теллура достаточно близки к другу и к экспериментальным значениям, а погрешность расчета методом ADF составляет не более 8 %.
Нами получены также достаточно надежные линейные корреляции (9-16), которые свидетельствуют о том, что проведенные расчеты галогенидов свинца методами РМ3 и B3LYP/SDD правильно описывают не только геометрию, но и термодинамические параметры, такие как энтальпии (ΔНо) и энтропии (Sо) образования, энергии диссоциации (Dо) (табл. 2).
Таблица 2
Рассчитанные и экспериментальные [15] энтальпии образования, энтропии, энергии диссоциации галогенидов свинца, кДж/моль и Дж/моль.К
|
Соединение |
ΔHо, PM3 |
ΔHо, эксп. |
Sо, расч. |
Sо, эксп. |
-Dо, расч. |
-Dо, эксп. |
|
PbF2 |
-375 |
-435.1 |
293 |
292.7 |
484 |
388 |
|
PbCl2 |
-166 |
-174.0 |
318 |
317.2 |
388 |
304 |
|
PbBr2 |
-119 |
-104.4 |
346 |
339.4 |
353 |
262 |
|
PbI2 |
-18 |
-3.2 |
362 |
359.6 |
315 |
209 |
|
PbF4 |
-617 |
-1133.4 |
345 |
333.5 |
341 |
327 |
|
PbCl4 |
-259 |
-552.4 |
399 |
381.6 |
260 |
252 |
|
PbBr4 |
-109 |
-456.4 |
445 |
426.2 |
229 |
199 |
|
PbI4 |
56 |
-224.5 |
477 |
466.3 |
200 |
164 |
Необходимо отметить, что если энтропии образования ложатся на одну зависимость для соединений Pb(II) и Pb(IV), то зависимости между рассчитанными и экспериментальными энтальпиями образования и энергиями диссоциации наблюдаются отдельно для соединений свинца различной координации на основании обоих методов расчета в программах GAUSSIAN и ADF:
GAUSSIAN: So (эксп.) = -23 + 0.914S0 (расч.) r = 0.996; s = 5; n = 8 (9)
ADF: So (эксп.) = 10.6 + 0.97S0 (расч.) r = 0.998; s = 4; n = 7 (10)
Для Pb(II):
PM3: ΔH0 (эксп.) = 29 +1.23 ΔH0 (расч.) r = 0.999; s = 12; n = 4 (11)
GAUSSIAN: Do (эксп.) = -108 +1.04 D0 (расч.) r = 0.993; s = 11; n = 4 (12)
ADF: Do (эксп.) = -44 +1.54 D0 (расч.) r = 0.990; s = 13; n = 4 (13)
Для Pb(IV):
PM3: ΔH0 (эксп.) = -289 +1,35 ΔH0 (расч.) r = 0.997; s = 35; n = 4 (14)
GAUSSIAN: Do (эксп.) = -62 +1.14 D0 (расч.) r = 0.992; s = 11; n = 4 (15)
ADF: Do (эксп.) = -28 +0.83 D0 (расч.) r = 0.986; s = 14; n = 4 (16)
Рассчитанные обоими методами значения энергетического расстояния между занятыми и свободными молекулярными орбиталями (ΔЕ ВЗМО-НСМО) хорошо согласуются с шириной запрещенной зоны в кристаллических хлоридах, бромидах и иодидах свинца [5] с коэффициентом корреляции 0.999 и стандартным отклонением 0.02.
Для дальнейшего изучения физико-химических свойств нами была проведена оптимизация элементарной ячейки PbS, PbSe и PbTe методом РМ3. Рассчитанные длины связей Pb-S, Pb-Sе и Pb-Te составила 3.5, 3.57 и 3.45 Å, что близко к экспериментальным значениям 2.97, 3.06 и 3.23 Å, а постоянные решеток оказались равными 6.5, 7.1 и 6.75 Å, что также близко к экспериментальному значению 5.94 Å для решетки сульфида свинца [16]. Необходимо отметить, что проведенный нами расчет по программе FLAPW кристаллических халькогенидов PbS, PbSе и PbTe дал лучшие значения постоянных решеток в 6.01, 6.23 и 6.57 Å, что практически совпадает с экспериментальными значениями.
Одной из наиболее важных характеристик полупроводников служит величина запрещенной щели. Расчет в твердом теле PbS, PbSе и PbTe дал значения 0.5, 0.44 и 0.70 эВ в газовой фазе – 0.38, 0.38 и 0.28 эВ по расчетам GAUSSIAN и 0.85, 0.36 и 0.25 эВ в ADF. Экспериментальная ширина зоны монокристалллического PbS равна 0.41 эВ [5]. Таким образом, можно предполагать, что проведенные расчеты газовой фазы и кристалла халькогенидов свинца дали достаточно близкие к эксперименту значения энергетических характеристик.
На основании расчета в твердом теле (FLAPW) и в газовой фазе методом ADF можно оценить химическую связь с точки зрения ионной (электростатической) и ковалентной (орбитальной) составляющей. Данные FLAPW указывают на смешанный ионно-ковалентный тип (рис. 4).
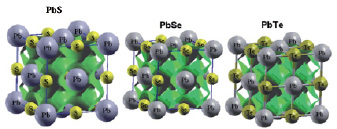
Рис. 4. Изоэлектронные поверхности (ρ = 0.15 e/Å3) кубических (типа В1) PbS, PbSe и PbTe
При этом ионная составляющая создается за счет зарядовой поляризации в направлении Pb→ S, Pb→ Sе и Pb→ Те. В идеальной ионной модели зарядовые состояния Pb2+Hal2-, в реальности эффективные заряды атомов-компонентов будут меньше за счет ковалентного вклада. Ковалентная составляющая образуется за счет частичного перекрывания Halnp – Pb6s,6р состояний. Ковалентную составляющую можно иллюстрировать рис. 4, где видно перекрывание изоэлектронных поверхностей в направлении Pb-(S, Se,Te). В программе ADF энергия молекулы разделяется на несколько вкладов. Энергетические члены могут быть идентифицированы тремя главными компонентами химической связи, то есть отталкиванием Паули, электростатическим притяжением и ковалентным взаимодействием. Френкинг и соавторы [18] предложили, чтобы ∆Еэлстат. использовался для оценки силы электростатического связывания, а ∆Еорб. – для ковалентного связывания. Наши результаты расчета халькогенидов свинца в газовой фазе также указывают на то, что химическая связь состоит из ионной и ковалентной составляющих с преобладанием последней (65 % для S, 79 % для Se и 56 % в случае Те).
Заключение
В работе показано, что использованный нами метод расчета B3LYP/SDD дает адекватные результаты в предсказании геометрических параметров, энергий диссоциации, ИК- и Раман-спектров соединений серы, селена и теллура. Рассчитанные методами FLAPW в твердом теле и B3LYP/SDD в газовой фазе длины связи и постоянные решетки халькогенидов свинца достаточно близки к экспериментальным значениям в сульфиде свинца. Проведен анализ ковалентной и ионной составляющих связей свинец-халькоген. Показано, что в газовой фазе и твердом теле больший вклад вносит ковалентная составляющая, уменьшающаяся от селена к теллуру.
Рецензенты:
Жерин И.И., д.х.н., профессор кафедры «Химическая технология редких, рассеянных и радиоактивных элементов» Физико-технического института Томского политехнического университета, г. Томск;
Ковалева С.В., д.х.н., профессор кафедры Томского государственного педагогического университета, г. Томск.
Работа поступила в редакцию 24.06.2014.
Библиографическая ссылка
Полещук О.Х., Егоров Н.Б., Полицинский Е.В., Ермаханов М.Н., Саидахметов П.А. КВАНТОВО-ХИМИЧЕСКОЕ ИССЛЕДОВАНИЕ СТРУКТУРЫ И СВОЙСТВ ХАЛЬКОГЕНИДОВ СВИНЦА // Фундаментальные исследования. 2014. № 9-3. С. 556-561;URL: https://fundamental-research.ru/ru/article/view?id=34886 (дата обращения: 22.07.2026).