



В настоящее время применение методов интеллектуального анализа данных в предметно-ориентированных информационных системах химических данных является актуальной научно-практической задачей [13]. Одним из таких методов является метод кластеризации, в результате которого могут быть построены группы химических объектов, близких по набору своих физико-химических характеристик. В ряде случаев построенное разбиение исходного множества на группы может быть использовано для предсказания некоторых характеристик объектов.
Целью настоящего работы является использование алгоритмов кластеризации к-средних для автоматического построения групп радикальных реакций отрыва, обладающих сходной реакционной способностью, по экспериментальным выборкам из нескольких баз данных.
Постановка задачи и метод решения
Элементарную радикальную реакцию отрыва атома водорода Rº + R1H → RH + Rº1 можно описать следующим набором характеристик [3, 6]:
1. Классическая энтальпия реакции:
DHe = Di – Df + 0,5(hLni – hLnf), (1)
где ni – частота валентного колебания разрываемой связи; nf – частота валентного колебания образующейся связи; h – постоянная Планка; L – число Авогадро; Di – энергия диссоциации разрываемой связи; Dei = Di + 0,5hLni,, где Df – энергия диссоциации образующейся связи, Def = Df + 0,5hLnf.
2. Классический потенциальный барьер Ee:
Ee = Ei + 0,5(hLni – RT), (2)
который отсчитывается от минимума потенциальной кривой рвущейся связи до точки минимума переходного состояния. R – газовая постоянная; T – абсолютная температура в К; Ei – экспериментальная энергия активации радикальной реакции.
3. Коэффициенты
b = p(2mi)1/2ni и bf = p(2mf)1/2nf, (3)
которые описывают зависимость потенциальной энергии от амплитуды колебаний атомов вдоль рвущейся (i) и образующейся (f) валентной связи, 2b2 – силовая постоянная связи, mi – приведенная масса атомов для разрываемой связи, mf – приведенная масса атомов для образующейся связи.
4. Коэффициент
a = b/bf . (4)
Первая задача исследования состоит в формировании метрического пространства признаков, исходя из характеристик радикальной реакции отрыва, и затем разбиении экспериментальной выборки реакций на группы в этом пространстве.
Введем переменные
X = (α2(Ee – ΔHe))1/4 и Y = (Ee)1/4.
Тройка {Rº + R1H → RH + Rº1, X, Y} будет метрическим пространством признаков для разбиения исходного набора радикальных реакций на группы (кластеры).
Источником экспериментальных послужили базы данных по константам скорости радикальных жидкофазных реакций и по энергиям диссоциации связей органических молекул предметно-ориентированной системы научной осведомленности в области физической химии радикальных реакций [7]. Для решения задачи группировки радикальных реакций был выбран массив экспериментальных данных по реакциям отрыва атома водорода алкильными радикалами от молекул углеводородов и их производных, выборка из базы данных по энергиям диссоциации связей органических соединений (269 реакций). Согласно классификации радикальных реакций, построенной экспертным путем [3, 6], выборка включала реакции четырех классов: 39 реакций с углеводородами и 100 реакций с производными от углеводородов, объединенных в класс Rº + R1H, 31 реакцию класса Rº + R2H, 99 реакций класса Rº + R3H, где Rº – алкильный радикал, R1H – углеводород, R2H – олефин, R3H – алкилароматический углеводород.
В качестве алгоритмов кластеризации были взяты классический алгоритм к-средних и алгоритм с-средних [8], для которых были разработаны соответствующие программы. Метрикой в алгоритмах кластеризации было евклидово расстояние.
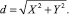 (5)
(5)
Были выполнены расчеты с различным числом кластеров для обоих алгоритмов (10, 20, 30 кластеров). Результаты разбиения на кластеры, полученные с использованием алгоритма к-средних и алгоритма с-средних, существенно не отличались за исключением того, что в нечетком случае наблюдалось смещение центра кластеров отношению классического случая.
Полученные в результате разбиения кластеры реакций в целом обладают следующими свойствами: кластер образует реакции либо одного радикала с группой соединений одного класса, либо реакции радикалов, которые имеют близкие значения образующей связи Df. Не наблюдается соответствия между классификацией радикальных реакций, построенной экспертным путем [3, 6] и полученной в результате кластеризации: имеет место, например, единичное попадание спиртов, простых и сложных эфиров в кластер с подавляющем содержанием алкилароматических соединений и т.п.). За недостаточностью места мы не будем приводить таблицу с полным разбиением. Рассмотрим несколько характерных примеров кластеров.
Так, кластер 1 содержит 8 реакций алкилароматических соединений с метильным радикалом, для которого Df = 440 кДж/моль [12] (показаны продукты реакции): {5 реакций CH4 + C6H5Cº(CH3)2, реакцию CH4 + (C6H5)2CºH и 2 реакции CH4 + C6H5CºH2}.
Кластер 3 содержит 3 реакции. Прочность С-Н-связи в CHCl3 Df = 392,5 кДж/моль, а в (CH3)3CH она составляет 400 кДж/моль [12]: {реакцию CHCl3 + цикло-[CºH(CH2)4], реакцию CHCl3 + (CH3)3Cº и реакцию (CH3)3CH + цикло-[(CH2)5CºH]}.
Кластер 7 содержит 5 реакций метильного радикала с циклоолефинами (Df = 440,0 кДж/моль [12]). {3 реакции CH4 + цикло-[CH = CHCºH(CH2)3], реакцию CH4 + цикло-[CH = CHCºH(CH2)4], реакцию CH4 + цикло-[CH = CHCºH(CH2)5]}.
Кластер 27 содержит 9 реакций алкильных радикалов с различными углеводородами. Для радикала CH3(CH2)9CºH2 прочность С-Н-связи Df = 422 кДж/моль [12] в CH3-группе. {2 реакции CH3(CH2)9CH3 + C6H5Cº(CH3)2, реакцию CH4 + CH2 = C(CH3)CºH2, реакцию CH4 + C6H5CºH2, 4 реакции CH4 + цикло-[C(O)CºH(CH2)3] и реакцию CH4 + CH3CºHC(O)CH2CH3}. В этом примере кластера алкильные радикалы атакуют различные углеводороды и их производные.
Таким образом, полученные результаты кластеризации отличаются от классификации, построенной экспертным путем. Кластеризация является более детализированной и более точно отражает структуру реакционного центра радикальной реакции отрыва.
Так же как и для классификации, полученной экспертным путем, с каждым кластером можно связать некоторое корреляционное соотношение, которое позволит делать предсказанное значений физико-химических характеристик реагентов [3]. При этом основная роль при предсказании реакционной способности молекул и энергий диссоциации разрываемой связи будет отведена координатам центров полученных кластеров.
Предсказание энергии диссоциации разрываемой С-Н-связи
Предсказание энергии диссоциации связи строится на предположении о том, что радикальная реакция отрыва с неизвестной разрываемой С–Н-связью попадает в определенный кластер. При этом предполагается, что 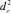 центра кластера и
центра кластера и 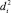 рассматриваемой реакции (неизвестно) отличаются не более чем на определенную величину. За такую величину можно принять
рассматриваемой реакции (неизвестно) отличаются не более чем на определенную величину. За такую величину можно принять
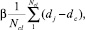 (6)
(6)
где Ncl –число элементов кластера; dj – расстояние j-элемента кластера; β – подгоночный параметр – положительная величина меньшая 0,5.
Тогда энергия диссоциации разрываемой С–Н-связи может быть вычислена с помощью следующих шагов.
Вычислить разность энергий активации реакций между реакцией-центром кластера и исследуемой реакции. В пределах одного кластера предэкспоненциальный множитель Ao в расчете на одну реагирующую связь остается постоянным, так что разность энергий активации реакций DEi с участием реагентов RсH и RiH может быть вычислена по формуле:
DEi = –RT ln((kin1)/(k1ni)), (7)
где ki – константа скорости исследуемой реакции; kс – константа скорости реакции-центра кластера; ni, nс – число эквивалентных атакуемых связей в исследуемой реакции и реакции-центра кластера соответственно.
Вычислить энтальпию опорной реакции:
DHeс = Diс1 – Dfс + 0,5(hLni – hLnf). (8)
Вычислить разность прочностей связей в реагенте и продукте. Из формулы расстояния, записанной для реакции-центра кластера и исследуемой реакции, вытекает следующая зависимость между разностью прочности связей DDi = Di – Df и DEi :
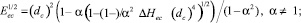 (9)
(9)
или
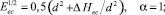 (10)
(10)
Eei = Eec + DEi; (11)
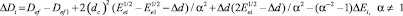 (12)
(12)
или
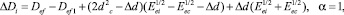 (13)
(13)
где Dd = (di)2 – (dc)2. В случае, когда атака в рассматриваемой паре реакций производится одинаковыми радикалами, то формула (11) принимает вид
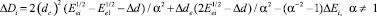 (14)
(14)
или
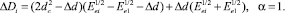 (15)
(15)
Вычислить энергию диссоциации разрываемой связи по формуле:
Di = DDi + D1 (16)
Покажем применение метода на практике. Рассмотрим кластер 7. Реакция метильного радикала с циклоалкенами. Центром кластера является реакция {метан + циклогексен} с d = 4.9. Прочность С-Н-связи в циклогексене в α-положении к двойной связи равна Df = 341,5 кДж/моль [12]. Результаты расчета приведены в табл. 1.
Как видно из таблицы, результаты вычислений находятся в хорошем согласии с данными справочника [12]: Di = 346,6; 357,2; 329,3 и 330,9 кДж/моль по строкам соответственно.
Предсказание классического потенциального барьера
Исходя из предположений, сделанных в предыдущем разделе статьи, оценить значение классического потенциального барьера исследуемой реакции можно по формулам:
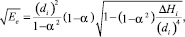 если α ≠ 1; (17)
если α ≠ 1; (17)
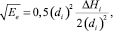 если α = 1. (18)
если α = 1. (18)
Таблица 1
Расчет энергий диссоциации связей Di в олефинах по кинетическим данным. Т – температура в К, ki – константа скорости реакции, k1 – константа скорости опорной реакции, Eei – энергия активации реакции, D1 – энергия диссоциации связи реакции – центра кластера, DDi – приращение энергии диссоциации связей, n1 и ni – число атакуемых связей в опорном и исследуемом реагентах
|
Радикал |
Реагент |
R1H Df, кДж/моль |
T |
ki/k1 |
n1/ni |
DEei |
DDi |
Di |
Л-ра |
|
CºH3 |
цикло-[CH = CHC{–H}(CH2)4] |
Циклогексен 341,5 |
350 |
0,63 |
1,00 |
1,60 |
5,08 |
346,9 |
[11] |
|
CºH3 |
цикло-[CH = CHC{–H}(CH2)5] |
Циклогексен 341,5 |
343 |
0,23 |
1,00 |
4,20 |
15,27 |
356,8 |
[11] |
|
CºH3 |
цикло-[CH = CHC{–H}(CH3)(CH2)2] |
Циклогексен 341,5 |
353 353 333 |
1,35 0,86 1,14 |
4,00 |
–4,95 –3,63 –4,18 |
–14,28 –10,38 –11,99 |
327,2 331,1 329,5 329,3 |
[1] [4] |
|
CºH3 |
цикло-[CH = CHCH = CHCH{–H}CH2] |
Циклогексен 341,5 |
333 |
1,93 |
2,00 |
–3,72 |
–10,04 |
330,9 |
[11] |
В табл. 2 приведен пример расчета по формуле (14) для уже рассмотренного нами кластера 27. Центром кластера является реакция {метан + кумол} с d = 4,2.
Таблица 2
Энергии активации реакций алкильных радикалов с C-H связью органических соединений Rº + RiH → RH + Riº при различных Т
|
RiH |
E (кДЖ/моль) |
|
|
CºH3 |
CH3(CH2)9CºH2 |
|
|
C6H5CH(CH3)2 |
16,8 (Т = 338 К) [10] |
26,7 (Т = 373 К) [5] |
|
C6H5CH3 |
28,2 (Т = 338 К) |
32,7 (Т = 373 К) |
|
CH2 = C(CH3)2 |
28,6 (Т = 338 К) [9] |
12,9 (Т = 373 К) |
|
цикло-[C(O)(CH2)4] |
28,9 (Т = 338 К) [2] |
49,8 (Т = 373 К) |
|
CH3CH2C(O)CH2CH3 |
23,4 (Т = 338 К) [2] |
51,5 (Т = 373 К) |
Полученные данные находятся в хорошем согласии с экспериментальными данными в пределах ошибки ±2,5 кДж/моль (для указанных литературных источников).
Заключение
В статье приведены результаты кластеризации экспериментальной выборки радикальных реакций отрыва типа Rº + RiH → RH + Riº алгоритмами к-средних. Показано, что полученное разбиение отличается от классификации, построенной экспертным путем.
На основе построенной кластеризации радикальных реакций отрыва предложен метод оценки энергии диссоциации С‒Н-связи по кинетическим и термохимическим данным для таких реакций с использованием координат центра кластера в построенном метрическом пространстве. Результаты полученного прогноза для контрольной выборки реакций находятся в хорошем согласии с экспериментальными данными.
В рамках построенной кластеризации предложен метод прогноза реакционной способности реакций радикального отрыва по термохимическим данным с использованием координат центра кластера.
Рецензенты:
Волохов В.М., д.ф.-м.н., зав. отделом, ИПФХ РАН, г. Черноголовка;
Психа Б.Л., д.х.н., зав. лаб., ИПХФ РАН, г. Черноголовка.
Работа поступила в редакцию 27.05.2013.
Библиографическая ссылка
Туманов В.Е. ПОСТРОЕНИЕ ГРУПП РАДИКАЛЬНЫХ РЕАКЦИЙ ОТРЫВА АЛГОРИТМОМ К-СРЕДНИХ ДЛЯ ПРЕДСКАЗАНИЯ ФИЗИКО-ХИМИЧЕСКИХ ХАРАКТЕРИСТИК РЕАГЕНТОВ // Фундаментальные исследования. 2013. № 6-6. С. 1386-1390;URL: https://fundamental-research.ru/ru/article/view?id=31746 (дата обращения: 02.08.2026).