


Scientific journal
Fundamental research
ISSN 1812-7339
"Перечень" ВАК
ИФ РИНЦ = 1,798
THE PLANNING AND VALIDATION STAGES OF QUALIFICATION OF CLEAN ROOMS IN THE PHARMACEUTICAL COMPANY

Согласно требованиям GMP для того, чтобы осуществлять контроль качества лекарственных средств, необходимо проводить валидацию процессов и квалификацию производственных систем. Под квалификацией производственных систем подразумевается квалификация оборудования, чистых помещений, инженерных систем [1].
Разработка производственной системы на фармацевтическом предприятии способствует повышению качества выпускаемой продукции, повышению эффективности деятельности предприятия в целом. Все валидационные и квалификационные мероприятия должны быть спланированы с учётом специфики производственных процессов, используемых оборудования и вспомогательных систем, а также специфики продукта [2]. Квалификационные и валидационные мероприятия необходимо выполнять в соответствии с установленными процедурами, квалифицированным персоналом, с соответствующим документальным оформлением. При этом привлекаются специалисты, которые обладают определённым опытом, имеют различные подходы и взгляды: пользователи, консультанты, поставщики, производственный персонал, персонал отдела обеспечения, контроля качества. Таким образом, при планировании валидации определяется объем испытаний и значимые аспекты, которые включают разработку мероприятий и плана валидации и квалификации [3].
Цель исследования: изучение этапов проведения квалификации, а также разработка схемы процесса проведения валидационных работ по квалификации функционирования чистых помещений.
Предмет квалификации: система вентиляции и кондиционирования чистых помещений.
Задача исследования: изучение методики протокола о проведении квалификации функционирования чистых помещений для документального подтверждения соответствия системы необходимым параметрам, нормам, нормативным документам и директивам по проектированию, строительству и эксплуатации чистых помещений.
Материалы и методы исследования
В процессе выполнения исследования использованы следующие методы и средства измерений.
1. Измерение расхода приточного воздуха и проверка кратности воздухообмена в чистом помещении: с помощью измерительного прибора (балометр) выполняется измерение потока воздуха за установленным воздушным фильтром в течение 6 с, а также форма и размер горловины измерительного прибора выбираются таким образом, чтобы они соответствовали размерам фильтра.
2. Проверка целостности и герметичности монтажа высокоэффективных воздушных фильтров. С помощью счетчика частиц проводится замер количества частиц, пришедших до HEPA-фильтра, и задается исходная концентрация (100 %). После чего проводится сканирование всей площади НЕРА-фильтра и фиксируется количество частиц, прошедших через него (как правило, скорость сканирования около 5 см/с, расстояние около 3 см от сканируемой поверхности). Тест герметичности установленной системы фильтрации проводится по всему периметру фильтра. После чего выполняется расчет процента проскока частиц.
3. Измерение счётной концентрации аэрозольных частиц. Счётчиком аэрозольных частиц проводятся измерения в точках согласно выполненному расчету и предоставленной планировке чистых помещений. Число точек пробоотбора и объемов пробы выбирается таким образом, чтобы были соблюдены требования ГОСТ ИСО 14644-1-2017 «Чистые помещения и связанные с ними контролируемые среды. Часть 1. Классификация чистоты воздуха». Измерения выполняются на уровне проведения технологических операций [4]. Точки отбора проб размещаются равномерно на высоте рабочей зоны или, если не обозначено иначе, на высоте 1,2 м от пола или площадки. Минимальное время измерения отбора проб составляет 1 мин. Минимальный объем отбора проб составляет 2 л. Если точка отбора проб в помещении одна, то в этой точке отбирается три пробы. Вычисляется средняя концентрация частиц по всем точкам отбора проб, замеренные значения числа частиц в м3 округляются до целого числа.
4. Измерение микроклиматических параметров чистых помещений (температура, относительная влажность, освещенность, шум). Измерения проводятся на высоте 1,5 м. Точки измерений равномерно распределяются по помещению. Число и размещение точек измерения принимается с учетом размера помещения или контролируемой зоны и мест, которые могут повлиять на измеряемые параметры (источники тепла, влажности, технологическое оборудование и т.п.).
Измерительный прибор помещают в точку измерения в тестируемом помещении на высоте рабочей зоны, и по истечении времени, необходимого для выхода измерительного прибора в стабильный режим, начинаются измерения. Измерения проводятся в течение 5 мин., показания прибора снимаются с интервалом в 1 мин. В каждой точке проводится не менее трёх измерений каждого параметра.
5. Контроль движения воздуха между участками (зонами), измерение перепадов давления.
6. Определение времени восстановления чистых помещений. Испытание выполняется для оценки способности чистых помещений восстанавливать заданный класс чистоты в течение определенного времени после внесения источника загрязнений. Испытание рекомендуется только для помещений с неоднонаправленным потоком воздуха. Определение времени восстановления происходит в соотношении 100:1. Проверка выполняется с введением искусственного аэрозоля. Как правило, проверка выполняется в одной точке. Для больших помещений из расчета по одной точке замеров на каждые 25 м2 поверхности пола.
Для выполнения работ по квалификации предложен пример протокола о квалификации функционирования чистых помещений.
Определим валидационную группу, которая будет отвечать за проведение квалификации чистых помещений. Ответственным является: начальник отдела валидации, инженер по валидации, специалист по валидации. Участники квалификационных работ несут ответственность за: сбор и анализ документации, необходимой для подготовки программы и протокола квалификации; разработку программы и протокола квалификации; выполнение работ по квалификации в соответствии с протоколом квалификации; подготовку результатов проведения квалификации в форме отчета по квалификации; выдачу заключений и рекомендаций по результатам квалификации.
Во время работы в рамках квалификации необходимо: подготовить протокол квалификации; согласовать протокол квалификации; выполнить тестирование; обработать данные измерений; проанализировать результаты квалификации функционирования; принять решения по выявленным отклонениям; разработать отчет о квалификации; согласовать и утвердить отчет о квалификации.
При проведении квалификации должны выполняться следующие условия: система должна находиться в состоянии покоя, т.е. система вентиляции и кондиционирования должна работать в штатном режиме на полную мощность, технологическое оборудование должно находиться в состоянии покоя, а в помещениях отсутствовать производственный персонал. Система должна быть очищена и отрегулирована. Во время проведения квалификации функционирования не должно проводиться вмешательство в работу оборудования и не должны проводиться изменения в наладке системы [5].
Результаты исследования и их обсуждение
Установлены основные этапы проведения квалификации, позволяющие в результате получить необходимые данные для подтверждения соответствия заданным параметрам.
Первым этапом является измерение расхода приточного воздуха и проверка кратности воздухообмена в чистом помещении.
При определении расхода приточного воздуха (производительности) выполняется измерение потока воздуха за установленным воздушным фильтром, полученный результат сравнивается с проектной документацией.
Кратность воздухообмена рассчитывается по формуле:
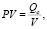
где PV – кратность воздухообмена помещения, м3/ч;
Qc – количество приточного воздуха, поступающего через все HEPA-фильтры, м3/ч;
V – объем помещения, м3.
Рассчитываемое значение сравнивается с проектным значением и дается ему оценка.
Вторым этапом является проверка целостности и герметичности монтажа высокоэффективных воздушных фильтров (НЕРА/ULPA): система вентиляции чистых помещений должна функционировать в нормальном режиме [6]. Распределительные решётки диффузоров должны быть удалены.
Третьим этапом является измерение счётной концентрации аэрозольныхчастиц.
Четвертым этапом – измерение микроклиматических параметров чистых помещений (температура, относительная влажность, освещенность, шум).
Пятым этапом – контроль движения воздуха между участками (зонами), измерение перепадов давления. Соседние помещения различных классов должны иметь перепад давления 10–15 Па. Перед началом теста валидационная группа должна определиться, по отношению к какому референтному давлению измеряется давление в комнате (атмосфера, окружающая среда квалифицируемого помещения). Во время измерения все квалифицируемые помещения должны быть закрыты. Измерение начинается после стабилизации давления. Прибор обнуляется, после чего к нему присоединяются гибкие трубки. Одна трубка вводится в референтную точку, вторая вводится в помещение, в котором проводится измерение. Размещение концов датчиков выбирается таким образом, чтобы на них не влияла близость приточно-вытяжной вентиляции помещения. Измеряются значения во всех смежных помещениях, в которые выходят двери, передаточные окна, стерилизационные туннели, технологические линии и т.п. – окружающая среда квалифицируемого помещения. Разность давлений измеряют дифференциальным манометром.
Шестым этапом – определение времени восстановления чистых помещений.
Для поиска возможных причин, которые могут повлиять на качество производства лекарственного препарата, установлены основные факторы, которые влияют на качество производства лекарственного средства на фармацевтическом предприятии. Причинно-следственная диаграмма Исикавы для выявления взаимосвязи основных факторов, влияющих на качество производимого лекарственного средства, представлена на рис. 1.
На основании выявленных факторов проведем оценку их влияния и определим наиболее значимые параметры, которые имеют решающее значение при производстве лекарственного средства. Результаты оценки наиболее значимых факторов, влияющих на производство лекарственного средства, представлены на рис. 2.
В ходе анализа наиболее значимых факторов, влияющих на производство лекарственного средства, установлено, что наиболее существенное влияние оказывают конструкция чистых помещений, монтаж системы вентиляции и кондиционирования.
На основании данного анализа можно сделать вывод, что актуальным является изучение основных этапов проведения квалификации чистых помещений и разработка формы протокола о проведении квалификации функционирования чистых помещений.
Таким образом, протокол о квалификации функционирования чистых помещений включает в себя следующее содержание:
– предмет квалификации функционирования; цель квалификации функционирования;
– идентификация системы (описание объекта квалификации);
– валидационная группа и распределение ответственности;
– режим работы в рамках квалификации; условия тестирования;
– критические операции, параметры и элементы системы;
– этапы проведения квалификации и критерии приемлемости; отчеты отклонений.
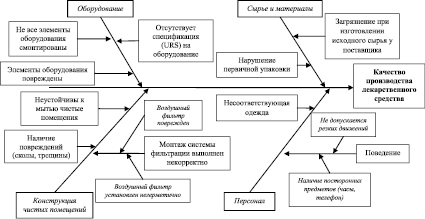
Рис. 1. Диаграмма Исикавы для выявления взаимосвязи основных факторов, влияющих на качество производства лекарственного средства
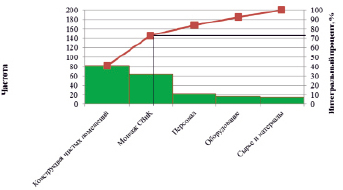
Рис. 2. Оценка основных факторов, влияющих на производство лекарственного средства
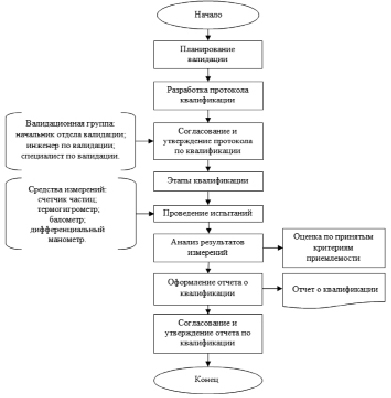
Рис. 3. Схема процесса проведения валидационных работ по квалификации функционирования чистых помещений
На основании проведенной квалификации составляется заключительный отчет, форма которого представлена в протоколе квалификации.
Схема процесса проведения валидационных работ по квалификации функционирования чистых помещений с использованием протокола представлена на рис. 3.
Выводы
Для повышения качества лекарственных препаратов, изготавливаемых на фармацевтическом предприятии, рационально создание производственной системы и проведение валидации процессов и квалификации производственных систем. Валидация содержит этапы проведения квалификации, проводимые на предприятии, такие как измерение расхода приточного воздуха и проверка кратности воздухообмена, проверка целостности и герметичности монтажа высокоэффективных воздушных фильтров, измерение счетной концентрации частиц, измерение микроклиматических параметров, измерение перепада давления между помещениями, определение времени восстановления. Таким образом, в процессе анализа подробно изучены методы испытаний, а также предложена методика проведения квалификации чистых помещений на фармацевтическом производстве.
Библиографическая ссылка
Татарникова А.А., Клейменова Н.Л., Назина Л.И., Орловцева О.А., Болгова И.Н. ПЛАНИРОВАНИЕ ВАЛИДАЦИИ И ЭТАПЫ ПРОВЕДЕНИЯ КВАЛИФИКАЦИИ ЧИСТЫХ ПОМЕЩЕНИЙ НА ФАРМАЦЕВТИЧЕСКОМ ПРЕДПРИЯТИИ // Фундаментальные исследования. 2020. № 4. С. 109-114;URL: https://fundamental-research.ru/en/article/view?id=42733 (дата обращения: 10.08.2026).
DOI: https://doi.org/10.17513/fr.42733