


Scientific journal
Fundamental research
ISSN 1812-7339
"Перечень" ВАК
ИФ РИНЦ = 1,798
STRUCTURAL DYNAMIC MODELS OF MONOGALOIDYS SUBSTITUTED SIX-MEMBERED CYCLIC COMPOUNDS IN THE CONDENSED STATE

Согласно современным научным воззрениям основные физико-химические свойства молекулярных объектов определяются формой их адиабатического потенциала. Под адиабатическим потенциалом в квантовой механике молекул принято считать результат решения квантового уравнения для электронной подсистемы исследуемого соединения [1]. Аналитическое решение такого уравнения не представляется возможным, что и привело к появлению упрощенных квантовых моделей для ансамбля электронов молекулярной системы. Созданные модели и стали основой такого известного научного направления в теории строения молекул, как квантовая химия, синонимом которой является квантовая теория молекул. В ней речь идет об использовании квантовой химии для построения структурно-динамических моделей молекулярных объектов, предсказания их конформационных и оптических свойств, спектроскопической интерпретации эксперимента по спектрам инфракрасного поглощения и комбинационного рассеяния.
Построение структурно-динамических моделей молекулярных соединений является одной из задач такого зародившегося современного научного направления, как молекулярное моделирование. Основы молекулярного моделирования заложены результатами хорошо развитой классической теории колебаний многоатомных молекул. Оценка гармонических параметров адиабатического потенциала молекулярных соединений различных классов базировалась на решении обратных спектральных задач. Для этого требовалось наличие полной информации по экспериментальным данным в спектрах ИК и КР и исходное нулевое поле силовых констант для исследуемого соединения. За подробностями отсылаем к широко известной в мировой науке монографии [2]. В монографии представлена обширная база данных по экспериментальной и теоретической интерпретации фундаментальных колебаний обширного списка соединений, их геометрии из рентгеноструктурного и электронографического эксперимента. Приведены и оценочные данные по гармоническим силовым константам (гармоническим параметрам адиабатического потенциала) для ряда молекулярных фрагментов.
Решение обратных спектральных задач требует наличия экспериментальной интерпретации спектра фундаментальных колебаний для изотопозамещенных аналогов исследуемого молекулярного объекта. Выбор же исходного приближения для системы гармонических силовых констант, варьирование которой до хорошего совпадения результатов теоретических расчетов колебательных состояний с данными эксперимента, требовал физического обоснования. Таковым была схема переноса гармонических силовых констант из родственных по электронной структуре молекулярных фрагментов.
Наглядным примером может служить теоретическая интерпретация колебательного спектра для шестичленных азациклов (пиридина, пиримидина, пиразина, пиридазина, триазина, тетразина), предложенная в монографии [2] на основании критического обзора соответствующей периодической литературы, вызванного расхождением в оценках частот фундаментальных колебаний азациклических соединений по данным эксперимента в ИК и КР спектрах. В качестве исходного силового поля использовались оценки гармонических силовых констант для молекулы бензола. Здесь известной трудностью являлась неоднозначность в выборе знака переносимых значений недиагональных силовых констант.
Имеющие известные недостатки классического подхода в оценке параметров адиабатического потенциала молекулярных соединений [2] послужили основанием для применения квантово-химических методов в обосновании полученных результатов. Исторически таковыми сначала являлись полуэмпирические методы, с помощью которых можно было устранить возникающие разногласия в схеме переноса гармонических силовых постоянных.
Активное развитие неэмпирических и гибридных квантовых методов молекулярной физики для исследования формы адиабатического потенциала, компьютерная реализация указанных методов в виде сервисных информационных технологий стали основой современного молекулярного моделирования. Появилась реальная возможность осуществления предсказательных расчетов структуры и спектров сложных молекулярных соединений в свободном и конденсированном состояниях, выявления признаков спектроскопической идентификации позиционных таутомеров и их возможных конформеров для исследуемого класса соединений, оценки значений гармонических и ангармонических параметров адиабатического потенциала. Квантовые методы стали использовать для устранения имеющихся разногласий в интерпретации колебательных спектров ряда молекулярных соединений, полученных в рамках классической схемы. К таковым в первую очередь следует отнести соединения химического промышленного производства. Для примера сошлемся на интерпретацию спектров карбоновых кислот [4]. Квантовые методы моделирования адиабатических параметров позволили приступить к теоретической интерпретации молекулярных объектов, для которых классический подход не может быть применен хотя бы ввиду отсутствия полного экспериментального исследования полос изотопозамещенных аналогов в ИК и КР спектрах и надежной интерпретации спектров отдельных фрагментов, входящих в состав исследуемого соединения. В качестве примера сошлемся на такие известные природные соединения, как гидроксизамещенные флавона – флавоноиды [3]. Возможность построения структурно-динамических моделей наиболее значимых представителей указанного класса соединений продемонстрирована в публикациях [5–6].
Однако квантовый подход в оценке параметров адиабатического потенциала сложных молекулярных объектов и построение их структурно-динамических моделей требует обоснования достоверности получаемых результатов.
Основным аргументом принято считать хорошее совпадение результатов теоретической интерпретации колебательных спектров исследуемого объекта с имеющимися в наличии экспериментальными данными по спектрам ИК и КР. Дополнительными можно считать факт воспроизводства низкочастотных колебаний (колебаний внутреннего вращения [2]) и характер поведения параметров полос в различных спектральных диапазонах. В этом случае можно сделать выводы о конформационных особенностях молекулярной геометрии и взаимном влиянии силовых полей отдельных молекулярных фрагментов.
В данной публикации продемонстрируем схему возможности обоснования доверительности предсказательных модельных квантовых оценок параметров адиабатического потенциала и предлагаемой на их основе интерпретации спектра фундаментальных колебаний моногалоидозамещенных шестичленных циклических соединений.
Модельные квантовые расчеты и их обсуждение
Модельные квантовые расчеты параметров адиабатического потенциала моногалоидозамещенных бензола, бензохинона, гамма-пирона и тиопирона (рисунок) осуществлялись в рамках метода функционала плотности DFT/b3LYP с использованием базисных наборов 6-311G** b 6-311+** [10]. Такой подход позволил констатировать, что выбор базиса не имеет принципиального значения для построения структурно-динамических моделей указанных галоидозамещенных шестичленных циклических соединений. Смещение положения полос не превышает величины ~ 10 см–1, качественная оценка интенсивностей сохраняется.
Диапазоны изменения расчетных значений геометрических параметров шестичленного фрагмента в монозамещенных (в том числе и галоидозамещенных) бензола, бензохинона, гамма-пирона и тиопирона представлены в табл. 1. Эти данные дают основание констатировать, что изменение геометрических параметров по отношению к значениям длин валентных связей и валентных углов носит локальный характер.
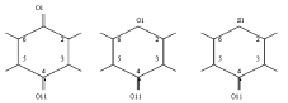
Нумерация атомов в бензохиноне, 4-гамма-пироне 4-тиопироне
Таблица 1
Расчетные значения геометрических параметров (ГП) монозамещенных бензола (МЗБ), бензохинона (МЗБХ), гамма-пирона (МЗГП) и тиопирона (МЗТП)
|
ГП |
МЗБ |
МЗБХ |
МЗГП |
МЗТП |
ГП |
МЗБ |
МЗБХ |
МЗГП |
МЗТП |
|
R (1, 2) |
1,39 |
1,50 |
1,34–1,36 |
1,72–1,75 |
A (2, 3, 8) |
118 |
121–122 |
115–120 |
118–119 |
|
R (1, 6) |
1,40 |
1,49 |
1,35–1,37 |
1,74 |
A (4, 3, 8) |
122 |
116 |
119–126 |
115–124 |
|
R (2, 3) |
1,39 |
1,34 |
1,34–1,35 |
1,35–1,36 |
A (3, 4, 5) |
121 |
117 |
112–113 |
116–117 |
|
R (3, 4) |
1,40 |
1,49 |
1,47–1,49 |
1,47–1,49 |
A (3, 4, 11) |
120 |
121 |
123–126 |
121–124 |
|
R (4, 5) |
1,40 |
1,48 |
1,47 |
1,46–1,48 |
A (5, 4, 11) |
120 |
122 |
122–124 |
119–122 |
|
R (5, 6) |
1,39 |
1,34 |
1,34 |
1,34–1,35 |
A (4, 5, 6) |
122 |
121 |
121–122 |
125–125 |
|
A (2, 1, 6) |
120 |
117 |
118 |
100–101 |
A (4, 5, 9) |
118 |
116 |
118–119 |
114–116 |
|
A (1, 2, 3) |
120 |
120 |
123–125 |
125–127 |
A (6, 5, 9) |
120 |
123 |
120 |
119 |
|
A (1, 2, 7) |
118 |
119–123 |
111–115 |
112–117 |
A (1, 6, 5) |
119 |
122–123 |
123–124 |
125–127 |
|
A (3, 2, 7) |
122 |
117–121 |
122–126 |
119–123 |
A (1, 6, 10) |
121 |
115 |
111 |
112–113 |
|
A (2, 3, 4) |
120 |
122–123 |
120–121 |
123–126 |
A (5, 6, 10) |
120 |
123 |
125 |
121–122 |
Примечание. Длины валентных связей R(i, i) в Å, значения валентных углов в A(i, j, k) в град.
В табл. 2–5 предложена интерпретация спектра фундаментальных колебаний бензола, бензохинона, гамма-пирона и тиопирона на основании квантовых оценок параметров адиабатического потенциала.
Таблица 2
Интерпретация спектра фундаментальных колебаний галоидозамещенных бензола
|
Форма колебаний |
С6Н5D |
С6Н5F |
С6Н5Cl |
С6Н5Br |
||||||||||
|
νэкс |
νвыч |
νэкс |
νвыч |
ИК |
КР |
νэкс |
νвыч |
ИК |
КР |
νэкс |
νвыч |
ИК |
КР |
|
|
Q, β, γ |
1591 |
1591 |
1603 |
1602 |
10 |
9 |
1583 |
1586 |
3 |
7 |
1585 |
1582 |
2 |
7 |
|
Q, β, γ |
1574 |
1574 |
1595 |
1598 |
46 |
8 |
1580 |
1582 |
25 |
15 |
1577 |
1577 |
26 |
15 |
|
β, Q |
1480 |
1480 |
1496 |
1487 |
80 |
1 |
1479 |
1471 |
47 |
0 |
1475 |
1467 |
42 |
0 |
|
β, Q |
1450 |
1450 |
1460 |
1450 |
2 |
0 |
1443 |
1440 |
7 |
1 |
1441 |
1437 |
8 |
1 |
|
β, Q |
– |
1321 |
1326 |
1311 |
0 |
0 |
1322 |
1310 |
0 |
1 |
1319 |
1309 |
1 |
1 |
|
β, Q |
1292 |
1292 |
– |
1290 |
0 |
1 |
1263 |
1286 |
0 |
0 |
1263 |
1284 |
0 |
1 |
|
qCX |
2269 |
2263 |
1220 |
1221 |
93 |
9 |
1068 |
1071 |
41 |
12 |
1071 |
1057 |
20 |
7 |
|
β |
1176 |
1176 |
1156 |
1151 |
0 |
2 |
1174 |
1169 |
0 |
2 |
1176 |
1171 |
0 |
3 |
|
β |
1158 |
1158 |
1156 |
1147 |
13 |
7 |
1157 |
1154 |
0 |
4 |
1158 |
1155 |
0 |
4 |
|
β, Q |
1076 |
1076 |
1065 |
1064 |
8 |
2 |
1083 |
1072 |
6 |
1 |
1068 |
1071 |
4 |
0 |
|
Q, β, γ |
1031 |
1031 |
1021 |
1015 |
4 |
8 |
1024 |
1019 |
20 |
20 |
1020 |
1014 |
24 |
28 |
|
γ |
1007 |
1007 |
1009 |
995 |
0 |
24 |
1002 |
992 |
6 |
24 |
1001 |
990 |
12 |
25 |
|
γ |
980 |
980 |
806 |
805 |
26 |
13 |
702 |
692 |
29 |
5 |
673 |
666 |
22 |
4 |
|
γ |
601 |
606 |
613 |
615 |
0 |
5 |
615 |
615 |
0 |
4 |
614 |
614 |
0 |
4 |
|
γ |
603 |
601 |
519 |
514 |
5 |
4 |
418 |
405 |
5 |
8 |
315 |
307 |
2 |
7 |
|
βCX |
858 |
858 |
407 |
396 |
2 |
0 |
279 |
292 |
0 |
1 |
254 |
245 |
0 |
1 |
|
ρ, χ |
995 |
995 |
997 |
968 |
0 |
0 |
987 |
980 |
0 |
0 |
990 |
979 |
0 |
0 |
|
ρ, χ |
970 |
970 |
970 |
949 |
0 |
0 |
965 |
956 |
0 |
0 |
964 |
955 |
0 |
0 |
|
ρ, χ |
922 |
922 |
896 |
886 |
8 |
1 |
903 |
896 |
2 |
0 |
905 |
897 |
2 |
0 |
|
ρ |
849 |
849 |
818 |
813 |
0 |
1 |
830 |
822 |
0 |
1 |
832 |
824 |
0 |
1 |
|
ρ, χ |
775 |
775 |
754 |
751 |
75 |
0 |
740 |
737 |
58 |
1 |
737 |
733 |
62 |
1 |
|
χ, ρ |
698 |
698 |
687 |
682 |
17 |
0 |
682 |
688 |
25 |
0 |
681 |
686 |
19 |
0 |
|
ρCX, χ |
608 |
608 |
519 |
501 |
10 |
0 |
470 |
470 |
11 |
0 |
460 |
459 |
9 |
0 |
|
χ |
405 |
405 |
407 |
416 |
0 |
0 |
418 |
409 |
0 |
0 |
409 |
406 |
0 |
0 |
|
χ |
380 |
380 |
242 |
235 |
0 |
4 |
196 |
184 |
0 |
2 |
181 |
164 |
0 |
3 |
Примечание. Частоты колебаний ν в см–1, интенсивности в спектрах ИК в моль/км, в спектрах КР в Å4/а.е.м.
Теоретическая оценка положения полос в колебательном спектре соединения осуществлялась с помощью известного соотношения ангармонической теории молекулярных колебаний:
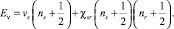 (1)
(1)
Для ангармонических констант χsr использованы выражения из публикации [8]:
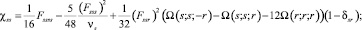 (2)
(2)
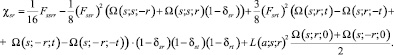 (3)
(3)
Отметим, что ангармоническое смещение полос, полученное с использованием приведенных соотношений, хорошо согласуется с результатами применения процедуры масштабирования типа [7]
νанг = (0,98 – 4,4Е – 0,6 νг) νг. (4)
Максимальное расхождение не превышает величины Δν ~ 20 см–1.
Для моногалоидозамещенных бензола (табл. 2) предложенная интерпретация спектра фундаментальных колебаний хорошо согласуется с результатами реального эксперимента, описанного в монографии [2]. Сравнение с интерпретацией колебаний для монодейтеробензола (С6H5D) дает основание сделать вывод, что влияние галоидозамещения на силовое шестичленного цикла носит локальный характер, что проявляется в характеристичности положения полос в диапазоне выше 1000 см–1. Существенное различие в значениях частот колебаний в диапазоне ниже 1000 см–1 есть результат влияния кинематических параметров заместителя (массы атома и длины связи СХ). Это относится в первую очередь к плоским деформационным колебаниям валентных углов шестичленного цикла (γ) и углов ССХ (βСХ).
Отметим характеристичность по частоте и интенсивности полос, отнесенных к неплоским деформационным колебаниям (ρ, χ) связей бензольного цикла. Смещение полосы (ρCX), отнесенной к выходу связи С-Х (D, F,Cl, Br), определяется в основном кинематическими параметрами заместителя Х. Следует обратить внимание на низкую интенсивность полос неплоских колебаний в спектрах КР. В спектре ИК в диапазоне 800–770 см–1 значительной интенсивностью обладает полоса, отнесенная к неплоскому колебанию типа ρ,χ.
Таблица 3
Интерпретация спектра фундаментальных колебаний галоидозамещенных 1,4-бензохинона (БХ)
|
Форма колебаний |
1,4-БХ |
1,4-БХ (d1) |
F-1,4-БХ |
Cl-1,4-БХ |
Br-1,4-БХ |
||||||||
|
νэкс |
νвыч |
νэкс |
νвыч |
νвыч |
ИК |
КР |
νвыч |
ИК |
КР |
νвыч |
ИК |
КР |
|
|
Qc = o, γ |
1686 |
1705 |
1679 |
1702 |
1711 |
225 |
100 |
1711 |
218 |
57 |
1707 |
219 |
43 |
|
Qc = o, γ |
1686 |
1686 |
1664 |
1685 |
1691 |
178 |
151 |
1690 |
162 |
164 |
1688 |
159 |
168 |
|
Q, β, γ |
1635 |
1630 |
– |
1624 |
1659 |
55 |
20 |
1636 |
2 |
24 |
1635 |
1 |
20 |
|
Q, β, γ |
1614 |
1600 |
1588 |
1589 |
1612 |
55 |
0 |
1591 |
92 |
20 |
1585 |
98 |
26 |
|
β, Q |
1394 |
1359 |
1363 |
1356 |
1349 |
6 |
16 |
1347 |
6 |
14 |
1345 |
7 |
13 |
|
β, Q |
1354 |
1354 |
1323 |
1307 |
1324 |
48 |
3 |
1286 |
19 |
6 |
1282 |
19 |
7 |
|
Q, β, γ |
1299 |
1259 |
1284 |
1250 |
1261 |
59 |
12 |
1251 |
102 |
12 |
1245 |
115 |
14 |
|
qCX |
3092 |
3094 |
2361 |
1178 |
77 |
1 |
972 |
133 |
1 |
951 |
109 |
1 |
|
|
β, Q |
1211 |
1190 |
1201 |
1171 |
1150 |
30 |
20 |
1171 |
3 |
21 |
1170 |
4 |
22 |
|
β, Q |
1147 |
1128 |
1100 |
1088 |
1071 |
94 |
9 |
1080 |
29 |
11 |
1075 |
41 |
12 |
|
γ, Q |
942 |
933 |
962 |
956 |
874 |
13 |
3 |
828 |
19 |
3 |
804 |
13 |
2 |
|
γ, Q |
770 |
752 |
765 |
746 |
770 |
1 |
1 |
763 |
1 |
2 |
760 |
1 |
3 |
|
γ |
728 |
726 |
– |
700 |
669 |
7 |
8 |
626 |
10 |
6 |
614 |
7 |
5 |
|
βC = O, γ, |
599 |
588 |
595 |
581 |
584 |
4 |
3 |
547 |
11 |
4 |
525 |
10 |
3 |
|
γ, βC = O |
447 |
448 |
– |
447 |
471 |
3 |
6 |
457 |
5 |
10 |
453 |
4 |
11 |
|
γ, βC = O |
447 |
443 |
445 |
435 |
428 |
8 |
4 |
422 |
20 |
2 |
419 |
25 |
1 |
|
γ, βC = O |
409 |
405 |
407 |
404 |
385 |
13 |
1 |
335 |
2 |
2 |
260 |
1 |
3 |
|
βCX |
1051 |
1057 |
875 |
859 |
301 |
0 |
1 |
227 |
0 |
3 |
183 |
0 |
3 |
|
ρ |
996 |
1007 |
988 |
1002 |
996 |
0 |
0 |
999 |
0 |
0 |
999 |
0 |
0 |
|
ρ, χ |
969 |
1000 |
933 |
949 |
888 |
75 |
0 |
907 |
53 |
0 |
909 |
52 |
0 |
|
ρ, χ |
882 |
893 |
850 |
856 |
832 |
5 |
4 |
831 |
19 |
2 |
827 |
18 |
3 |
|
χ, ρC = O |
793 |
785 |
792 |
783 |
761 |
0 |
6 |
772 |
0 |
5 |
770 |
0 |
5 |
|
χ, ρ |
747 |
748 |
670 |
678 |
613 |
3 |
1 |
591 |
0 |
3 |
583 |
0 |
2 |
|
ρCХ,χ |
505 |
508 |
474 |
478 |
404 |
0 |
0 |
386 |
2 |
0 |
375 |
1 |
0 |
|
χ |
310 |
335 |
300 |
325 |
270 |
0 |
2 |
259 |
0 |
2 |
255 |
0 |
2 |
|
χ |
244 |
228 |
238 |
224 |
170 |
2 |
1 |
149 |
3 |
1 |
139 |
3 |
1 |
|
χ |
89 |
93 |
108 |
92 |
90 |
9 |
0 |
83 |
9 |
0 |
80 |
8 |
1 |
Для галоидозамещенных бензохинона не располагаем экспериментальными данными, поэтому интерпретация спектра фундаментальных колебаний моногалоидозамещенных аналогов бензохинона (табл. 3) носит предсказательный характер. Обоснованием достоверности представленных результатов могут служить закономерности в характере поведения полос при монозамещении. Для монодейтеробензохинона (1,4-БХ (d1)) имеем хорошее совпадение квантовых модельных расчетов спектра фундаментальных колебаний с данными из реального эксперимента [11, 12]. Характер смещения полос при монозамещении по отношению к спектру базовой молекулы (1,4-БХ) может служить обоснованием достоверности предлагаемой интерпретации спектра фундаментальных колебаний моногалоидобензопиронов.
Проявление свойства характеристичности полос по частоте в диапазоне выше 1000 см–1 дает основание сделать вывод о локальном влиянии заместителей на силовое поле шестичленного циклического фрагмента. Смещение полос в диапазоне ниже 1000 см–1 обусловлено влиянием кинематических параметров заместителя. В первую очередь это касается колебаний, отнесенных по форме к типу γ, βCX, полосы которых характеризуются сравнительно малой интенсивностью в спектрах ИК и КР. Имеет место факт слабой интенсивности для большинства полос, отнесенных к неплоским деформационным колебаниям шестичленного циклического фрагмента моногалоидозамещенных бензохинона.
Предлагаемая интерпретация спектра фундаментальных колебаний моногалоидозамещенных гамма-пирона, представленная в табл. 4, является предсказательной. Располагаем только экспериментальными данными [9] для базового соединения – гамма-пирона (ГП).
Таблица 4
Интерпретация спектра фундаментальных колебаний галоидозамещенных гамма-пирона (ГП)
|
Форма колебаний |
ГП |
ГП-d2 |
2F-ГП |
2Cl-ГП |
ГП-d3 |
3F-ГП |
3Cl-ГП |
||||||||
|
νэкс |
νвыч |
νвыч |
ИК |
КР |
νвыч |
ИК |
КР |
νвыч |
νвыч |
ИК |
КР |
νвыч |
ИК |
КР |
|
|
Qc = o |
1700 |
1695 |
1701 |
499 |
47 |
1699 |
454 |
52 |
1691 |
1702 |
388 |
54 |
1706 |
338 |
38 |
|
Q, β, γ |
1637 |
1617 |
1664 |
141 |
15 |
1628 |
85 |
20 |
1624 |
1645 |
18 |
25 |
1628 |
35 |
41 |
|
Q, β |
1593 |
1561 |
1591 |
20 |
6 |
1566 |
46 |
9 |
1573 |
1584 |
21 |
10 |
1565 |
0 |
6 |
|
β |
1417 |
1386 |
1374 |
11 |
2 |
1375 |
20 |
2 |
1385 |
1388 |
15 |
0 |
1377 |
13 |
2 |
|
β, Q, γ |
1398 |
1355 |
1357 |
320 |
0 |
1321 |
297 |
0 |
1369 |
1361 |
13 |
4 |
1335 |
25 |
5 |
|
β |
1216 |
1236 |
1230 |
1 |
3 |
1223 |
27 |
6 |
1304 |
1292 |
48 |
2 |
1298 |
101 |
2 |
|
qCX |
3089 |
2318 |
1205 |
114 |
5 |
1047 |
136 |
4 |
2297 |
1227 |
164 |
5 |
1035 |
120 |
5 |
|
β |
1197 |
1198 |
1177 |
18 |
0 |
1189 |
1 |
1 |
1185 |
1161 |
99 |
4 |
1190 |
8 |
6 |
|
β, Q, γ |
1029 |
1067 |
1026 |
6 |
8 |
1013 |
10 |
11 |
1099 |
1094 |
6 |
2 |
1081 |
4 |
2 |
|
γ, Q |
1004 |
987 |
928 |
73 |
4 |
918 |
74 |
7 |
1002 |
968 |
34 |
12 |
951 |
82 |
8 |
|
γ, Q |
922 |
951 |
912 |
1 |
3 |
856 |
6 |
2 |
917 |
862 |
38 |
4 |
836 |
9 |
12 |
|
γ, Q |
790 |
841 |
743 |
7 |
16 |
699 |
12 |
11 |
760 |
722 |
8 |
11 |
666 |
11 |
6 |
|
γ |
474 |
491 |
532 |
6 |
4 |
501 |
2 |
4 |
488 |
577 |
4 |
7 |
538 |
9 |
7 |
|
γ, βC = O |
638 |
623 |
513 |
3 |
3 |
472 |
9 |
0 |
627 |
538 |
7 |
3 |
517 |
5 |
3 |
|
γ, βC = O |
453 |
444 |
481 |
6 |
1 |
405 |
8 |
8 |
436 |
440 |
1 |
1 |
373 |
0 |
3 |
|
βCX |
1319 |
880 |
360 |
8 |
1 |
258 |
2 |
3 |
851 |
290 |
1 |
1 |
225 |
1 |
2 |
|
ρ |
969 |
947 |
944 |
0 |
0 |
951 |
0 |
1 |
947 |
946 |
0 |
0 |
948 |
0 |
0 |
|
ρ, χ |
922 |
900 |
847 |
76 |
1 |
867 |
51 |
1 |
911 |
861 |
40 |
1 |
881 |
20 |
1 |
|
ρ |
857 |
775 |
811 |
3 |
2 |
818 |
17 |
2 |
846 |
831 |
35 |
2 |
835 |
49 |
1 |
|
ρC = O, χ |
790 |
734 |
707 |
1 |
3 |
709 |
1 |
3 |
765 |
737 |
0 |
3 |
740 |
0 |
3 |
|
ρCX, χ |
724 |
709 |
648 |
5 |
2 |
586 |
0 |
2 |
628 |
503 |
10 |
1 |
481 |
7 |
3 |
|
χ |
430 |
423 |
404 |
6 |
1 |
403 |
7 |
1 |
429 |
401 |
3 |
1 |
401 |
5 |
0 |
|
χ |
– |
364 |
202 |
0 |
1 |
160 |
2 |
0 |
388 |
296 |
1 |
1 |
253 |
0 |
0 |
|
χ |
– |
155 |
155 |
6 |
0 |
151 |
5 |
0 |
152 |
132 |
3 |
1 |
119 |
3 |
1 |
Таблица 5
Интерпретация спектра фундаментальных колебаний галоидозамещенных тиопирона (ТП)
|
Форма колебаний |
ТП d2 |
2F-ТП |
2Cl-ТП |
ТП-d3 |
3F-ТП |
3Cl-ТП |
||||||||
|
νвыч |
νвыч |
ИК |
КР |
νвыч |
ИК |
КР |
νвыч |
νвыч |
ИК |
КР |
νвыч |
ИК |
КР |
|
|
QC = O, γ |
1687 |
1685 |
425 |
66 |
1682 |
412 |
68 |
1683 |
1689 |
389 |
73 |
1691 |
316 |
52 |
|
Q, β, γ |
1590 |
1626 |
116 |
18 |
1595 |
64 |
24 |
1592 |
1608 |
21 |
32 |
1586 |
21 |
42 |
|
Q, β |
1548 |
1573 |
5 |
3 |
1554 |
36 |
3 |
1548 |
1564 |
17 |
4 |
1546 |
5 |
8 |
|
β, Q |
1350 |
1353 |
31 |
1 |
1354 |
41 |
1 |
1349 |
1370 |
16 |
0 |
1353 |
10 |
0 |
|
Q, β, γ |
1295 |
1287 |
60 |
1 |
1271 |
50 |
3 |
1333 |
1337 |
28 |
4 |
1316 |
15 |
6 |
|
β, Q |
1233 |
1213 |
15 |
1 |
1230 |
2 |
2 |
1215 |
1199 |
10 |
12 |
1207 |
10 |
10 |
|
β, Q |
1148 |
1149 |
30 |
12 |
1146 |
35 |
12 |
1127 |
1110 |
55 |
1 |
1124 |
12 |
3 |
|
qCX |
2345 |
1138 |
146 |
1 |
928 |
72 |
4 |
2352 |
1164 |
119 |
3 |
966 |
63 |
2 |
|
γ, Q |
949 |
892 |
2 |
3 |
839 |
36 |
0 |
973 |
885 |
27 |
0 |
828 |
44 |
1 |
|
γ, Qcs |
738 |
757 |
3 |
3 |
755 |
1 |
4 |
742 |
786 |
8 |
13 |
764 |
22 |
19 |
|
Qcs, γ |
674 |
703 |
20 |
1 |
698 |
23 |
3 |
705 |
738 |
6 |
2 |
730 |
20 |
2 |
|
γ, Q |
665 |
640 |
16 |
16 |
609 |
11 |
12 |
670 |
637 |
22 |
9 |
593 |
10 |
6 |
|
γ, βC = O |
552 |
478 |
2 |
8 |
449 |
12 |
3 |
553 |
538 |
8 |
4 |
517 |
14 |
2 |
|
γ, βC = O |
425 |
456 |
6 |
1 |
429 |
0 |
9 |
420 |
440 |
2 |
8 |
418 |
4 |
10 |
|
γ |
406 |
390 |
9 |
6 |
343 |
7 |
7 |
407 |
390 |
3 |
4 |
337 |
1 |
5 |
|
βCX |
848 |
324 |
7 |
0 |
230 |
3 |
2 |
858 |
306 |
1 |
0 |
236 |
1 |
2 |
|
ρ |
965 |
956 |
0 |
0 |
958 |
0 |
0 |
965 |
954 |
0 |
0 |
958 |
0 |
0 |
|
ρ, χ |
920 |
857 |
58 |
1 |
879 |
38 |
0 |
892 |
834 |
82 |
0 |
848 |
64 |
0 |
|
ρC = O, ρ |
812 |
788 |
12 |
3 |
787 |
21 |
2 |
828 |
803 |
2 |
3 |
807 |
9 |
1 |
|
ρ, χ |
719 |
644 |
19 |
2 |
637 |
16 |
3 |
697 |
679 |
11 |
3 |
677 |
9 |
3 |
|
ρCX, χ |
592 |
513 |
1 |
2 |
482 |
5 |
1 |
546 |
426 |
0 |
0 |
406 |
2 |
1 |
|
χ |
365 |
358 |
0 |
0 |
358 |
0 |
0 |
363 |
355 |
0 |
1 |
361 |
1 |
0 |
|
χ |
332 |
191 |
1 |
1 |
159 |
1 |
1 |
353 |
247 |
1 |
1 |
209 |
0 |
0 |
|
χ |
112 |
109 |
4 |
1 |
105 |
4 |
1 |
111 |
101 |
3 |
1 |
91 |
3 |
1 |
Согласно принятому в классическом подходе физическому предположению о сохранении значений параметров адиабатического потенциала при изотопозамещении [2], в табл. 3 предложена интерпретация спектра фундаментальных колебаний для 2-d и 3-d монодейтрозамещенных ГП. Влияние массы дейтерия определяет корреляцию частот колебаний по отношению к ГП, характер которой, предложенный в монографии [2], можно считать обоснованием достоверности отнесения для 2-d и 3-d монодейтрозамещенных ГП.
Обоснованием достоверности предлагаемого отнесения спектра фундаментальных колебаний для позиционных таутомеров (2-Х и 3-Х, X = F,Cl) моногалоидозамещенных ГП может служить характер корреляции частот колебаний по отношению к соответствующим позиционным таутомерам дейтерозамещенных ГП. Характеристичность по частоте имеет место для диапазона выше 1000 см–1, кинематические параметры заместителей определяют смещение полос в диапазоне ниже 1000 см–1. Проявляется и такая закономерность, низкая интенсивность полос, отнесенных к неплоским колебаниям моногалоидозамещенных ГП.
Для тиопирона (ТП) и таутомеров моногалоидозамещенных (ГП) не располагаем экспериментальными данными. Достоверность предсказательной интерпретации спектров, предложенной в табл. 5, может быть обоснована теми же аргументами, что выше выдвинуты для моногалоидозамещенных бензола, бензохинона, гамма-пирона.
Заключение
Сопоставление результатов теоретической интерпретации спектра фундаментальных колебаний рассмотренных моногалоидозамещенных шестичленных циклических соединений с имеющимися экспериментальными данными по спектрам ИК и КР дает основание для следующих выводов.
Метод функционала плотности DFT/b3LYP позволяет осуществлять доверительные предсказательные расчеты колебательных состояний монозамещенных циклических соединений при замещении атома углерода карбонового цикла на атомы кислорода и серы. Такой вывод в периодической литературе был сделан для ряда азациклических шестичленных соединений.
Монозамещения оказывают локальное влияние на силовое поле шестичленного цикла соединений, что проявляется в свойствах характеристичности по частоте фундаментальных колебаний в диапазоне выше 1000 см–1, а также для ряда неплоских деформационных колебаний рассмотренных молекулярных объектов. Кинематические свойства заместителей определяют смещение полос в диапазоне ниже 1000 см–1, отнесенных к плоским деформационным колебаниям валентных углов шестичленного цикла. Интенсивность указанных колебаний в спектрах ИК и КР незначительна. В этот же диапазон попадают и полосы неплоских деформационных колебаний связей СН, С = О, СХ. Большинство из них обладает низкой интенсивностью. Особенно это касается спектров КР.
Полученные результаты могут иметь и практическое значение для обоснования предсказательных расчетов монозамещенных шестичленных циклических соединений, полосы фрагменты-заместители которых проявляются в диапазоне 1600–1200 см–1, что проявляется в факте делокализации по форме для фундаментальных колебаний. В галоидозамещенных шестичленных циклических соединениях такого факта не наблюдается, а в амино-, нитро-, гидрокси-, карбокси-, альдегидо- и более сложных заместителей вызывает трудности в задаче построения структурно-динамических моделей и выделения признаков спектроскопической идентификации возможных позиционных таутомеров и их конформеров.
Рецензенты:
Булатов М.Ф., д.ф.-м.н., профессор, генеральный директор «Гиредмет», г. Москва;
Русаков В.С., д.ф.-м.н., профессор кафедры общей физики, МГУ, г. Москва.
Библиографическая ссылка
Алыкова О.М., Смирнов В.В., Элькин М.Д., Безниско Е.И. СТРУКТУРНО-ДИНАМИЧЕСКИЕ МОДЕЛИ МОНОГАЛОИДОЗАМЕЩЕННЫХ ШЕСТИЧЛЕННЫХ ЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ В КОНДЕНСИРОВАННОМ СОСТОЯНИИ // Фундаментальные исследования. 2015. № 2-21. С. 4635-4642;URL: https://fundamental-research.ru/en/article/view?id=38042 (дата обращения: 31.07.2026).