


Scientific journal
Fundamental research
ISSN 1812-7339
"Перечень" ВАК
ИФ РИНЦ = 1,798
CLINICAL AND GENETIC ASPECTS OF THE ARTERIAL HYPERTENSION IN CONDITION OF THE CHRONIC STRESS

Артериальная гипертензия (АГ) является одним из наиболее распространенных заболеваний в мире [9]. Гипертония диагностирована у 37–55 млн жителей Европы [15]. АГ выступает основным сердечно-сосудистым фактором риска развития ишемического инсульта, ишемической болезни сердца, причиной увеличения общей смертности населения [1]. Многообразие патогенетических механизмов, участвующих в развитии АГ, и обусловливает полиморфность ее клинической картины. Наряду с нейрогуморальной, ренальной и другими концепциями патогенетических механизмов АГ, важное место отводится генетическим факторам [2, 7]. С позиций генетики данная патология характеризуется клинико-генеалогической неоднородностью, мультифакториальностью проявлений и наследования. Вместе с тем использование молекулярно-генетических методов, направленных на выявление и оценку генетического риска, прогнозирование осложнений заболевания является перпективным для дальнейшего изучения влияния генов-кандидатов на прогрессирование АГ.
При АГ изменяется морфофункциональное состояние стенки артерии, главным образом за счет развития эндотелиальной дисфункции (ЭД) [3, 4, 5]. ЭД связана с нарушением баланса медиаторов, обеспечивающих сосудистый тонус, в первую очередь, со снижением биодоступности оксида азота (NO). Это конкурентное подавление и снижение активности эндотелиальной NO-синтазы за счет эндогенного L-аргинина, который используется в неокислительной аргиназной реакции. Другой механизм развития ЭД – инактивация NO за счет увеличения продукции активных форм кислорода, таких как супероксид-анионы и др. [6]. Независимо от диаметра просвета и площади поперечного сечения стенки сосуда происходит утолщение комплекса интима-медиа (КИМ). В крупных артериях наблюдается увеличение образования коллагена и уменьшение содержания эластина в медии, что сопровождается снижением эластических свойств [1, 2].
В связи с этим интерес вызывает изучение генетических детерминант ЭД у больных АГ. Одним из генов, роль которого в развитии АГ и поражении органов-мишеней широко обсуждается, является ген эндотелиальной NO-синтазы (eNOS) [4, 14, 13]. Значение этого гена подтверждается экспериментальным исследованием: у мышей с разрушенным геном eNOS отмечали более высокие цифры АД, чем в контрольной серии [2, 3, 7].
Оксид азота (NO) – основной эндотелиальный фактор релаксации гладкомышечной мускулатуры сосудов, участвующий в поддержании тонуса сосудистой стенки. NO синтезируется из L-аргинина с помощью семейства ферментов NO-синтетаз. NO участвует в патогенезе ишемической болезни сердца за счет способности NO угнетать пролиферацию гладкомышечных клеток, протективного эффекта в отношении агрегации тромбоцитов, свойству ингибировать адгезию лейкоцитов к эндотелию [11, 13]. Снижение активности eNOS приводит к недостатку NO и, как следствие, к ЭД, которой, согласно классической теории «ответ на повреждение», отводится основная роль в инициации атерогенеза [10, 12].
Целью работы явилось изучение роли полиморфизма Т-786С промотора гена eNOS в развитии АГ в условиях действия хронического стресса (ХС).
Материалы и методы исследования
В качестве объекта, подверженного воздействию ХС, обследовали 141 машиниста магистральных локомотивов (ММЛ). Сформировано 5 групп ММЛ в зависимости от возраста и стажа работы (СР). При этом СР выступал в качестве меры длительности воздействия ХС. 1 группу составили 28 ММЛ после окончания техникума, возраст 19,12 ± 0,89 (СР до 1 года); 2 группу – 28 ММЛ, возраст 27,54 ± 1,18 (СР 5–7 лет); 3 группу – 29 человек, возраст 37,41 ± 1,09 (СР 14–17 лет); 4 группу – 28 ММЛ, возраст 46,37 ± 1,06 (СР 21–24 года) и 5 группу – 28 человек, возраст 56,51 ± 1,02 (СР 30–34 года).
Динамику среднесуточного артериального давления (АД) в группах изучали методом холтеровского мониторирования АД с помощью аппарата «Кардиотехника 04», производства ИНКАРТ, Россия.
Толщину КИМ измеряли на ультразвуковом допплеровском аппарате VІVІD-3, компании GE (США), в режиме триплексного сканирования, датчиком 7 МГц. Измерение проводили в области задней стенки общей сонной артерии (ОСА) на расстоянии 1 см от ее бифуркации.
Cодержание ЭТ-1 определяли иммуноферментным методом в сыворотке крови с использованием реагентов фирмы DRG (США).
Исследовали полиморфизм T-786C промотора гена эндотелиальной NO-синтазы. Ген, кодирующий eNOS, находится в хромосоме 7q35–36 и состоит из 26 экзонов [11]. Промотор гена eNOS содержит несколько доменов, то есть, может регулироваться рядом факторов транскрипции [9, 11]. На сегодня описан полиморфизм гена eNOS в 11 местах, 8 из которых изучены в качестве возможных факторов риска сердечно-сосудистых заболеваний [8, 10].
Изучение аллельного полиморфизма Т-786С промотора гена eNOS проводили в отделе молекулярно-генетических исследований Центральной научно-исследовательской лаборатории Донецкого национального медицинского университета им. М. Горького. ДНК выделяли из цельной крови с помощью реагента «Проба-рапид генетика» (ДНК-Технология, Россия). Анализ полиморфных ДНК-локусов выполняли методом полимеразной цепной реакции (ПЦР) с помощью тест-системы «SNP-экспресс: Т-786С промотора гена eNOS» (НПФ Литех, Россия). ПЦР проводили на амплификаторе «GeneAmp PCR System 2400» (CША). Детекцию амплифицированных фрагментов осуществляли путем электрофореза в 3 %-м агарозном геле, окрашенном бромистым этидием, с последующей визуализацией в ультрафиолетовом трансиллюминаторе «TFX-20.M» («Vilber Lourmat», Франция).
Статистические расчеты осуществляли с применением программы «STATISTICA 6.1» (StatSoft, Inc.). Достоверность различий в распределении генотипов между группами, а также соответствие распределения закону Харди ‒ Вайнберга оценивали с помощью анализа таблиц сопряженности по критерию χ2. Степень ассоциации генотипа с наличием АГ рассчитывали по величине отношения шансов (ОR) с учетом 95 % доверительного интервала (ДИ). Влияние вариантов генного полиморфизма на клинико-функциональные показатели устанавливали с использованием дисперсионного анализа. Достоверность различий проводили на уровне значимости 5 % с использованием t-критерия Стьюдента.
Результаты исследования и их обсуждение
Результаты среднесуточного мониторирования АД (СМАД) показали, что уже начальный период действия стрессоров сопровождается ростом цифр АД у ММЛ. При этом отмечается тенденция к увеличению количества ММЛ с АГ в зависимости от стажа работы (рис. 1).
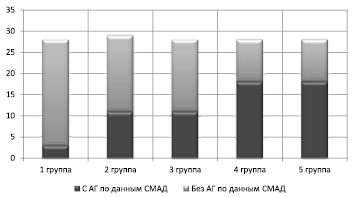
Рис. 1. Распределение обследованных ММЛ в зависимости от длительности воздействия ХС и наличия/отсутствия признаков АГ
Длительность влияния стрессоров определяло тяжесть формирующейся АГ: с увеличением сроков действия ХС усиливалась степень тяжесть АГ, в первую очередь это происходило за счет сокращения количества ММЛ с нормальными значениями АД (рис. 2).
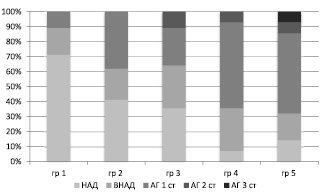
Рис. 2. Степень тяжести АГ у ММЛ в зависимости от длительности действия ХС: НАД – нормальное АД; ВНАД – высокое нормальное АД
Генетические факторы занимают особое место в процессах ремоделирования сосудистой стенки, так как их влияние может реализовываться при участии как гемодинамических, так и нейрогуморальных механизмов. Для оценки роли генетических факторов в развитии АГ в условиях действия ХС машинисты были разделены на две группы. В первую группу вошел 61 ММЛ с различной степенью тяжести АГ. Вторую группу – контрольный контингент (КК) – составили 50 ММЛ с нормальными цифрами АД.
Результаты генетических исследований Т-786С промотора гена eNOS у ММЛ с АГ показали следующее соотношение генотипов: ТТ – 16,4 %, ТС – 50,8 % и СС – 32,8 %. В группе КК это соотношение составило 48; 46 и 6 % (рис. 3). Наблюдаемое распределение частоты выявляемости генотипов и аллелей гена eNOS в группе обследованных ММЛ с наличием АГ и в группе КК соответствовало равновесию Харди – Вайнберга.
Таким образом, в группе ММЛ с АГ примерно в 5,5 раза чаще, чем в группе КК, выявляли гомозиготы с генотипом СС промотора гена eNOS (соответственно 32,8 и 6,0 %, p(F) < 0,001). Полученные данные позволяют предположить патогенетическое значение данного полиморфизма в развитии АГ в условиях действия ХС. По данным экспериментальных исследований [10, 13], наличие аллеля С в положении 786 промотора гена eNOS приводит к снижению активности фермента eNOS вполовину, а формирующийся в результате этого его недостаток является причиной снижения синтеза и высвобождения оксида азота и дисфункции эндотелия. Снижение синтеза NO сопровождается нарушением тонической вазодилатации сосудов, приводит к преобладающему влиянию вазоконстрикторных регуляторов и стойкому вазоспазму как одному из патогенетических звеньев формирования АГ. Доказано, что у людей с генотипом СС и ТС промотора гена eNOS увеличен тонус коронарных артерий и повышена склонность к коронароспазму [4, 14].
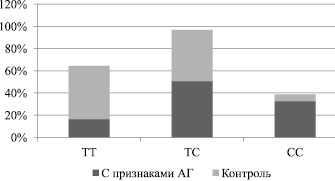
Рис. 3. Полиморфизм Т-786С промотора гена eNOS у ММЛ с признаками АГ и в группе КК
При анализе генотипических и фенотипических особенностей ММЛ с АГ установлено, что риск развития АГ при гомозиготном варианте СС (odds ratio (OR) = 7,6 (2,12 – 27,6), р < 0,01) выше, чем при гетерозиготном варианте ТС ((OR) = 1,2 (0,573 – 2,566), р = 0,133). Генотип СС является независимым фактором риска развития АГ. В ходе проведенного исследования показано (табл. 1), что сам факт наличия аллеля С является фактором риска развития АГ (OR = 3,4 (1,9 – 5,98), р = 0,0001) при сравнении с аллелем Т.
Проведенное исследование показало, что у ММЛ с наличием АГ КИМ ОСА претерпевает более выраженные изменения, чем у ММЛ с нормальными значениями АД. Так, разница толщины КИМ ОСА у ММЛ с признаками АГ и нормальным АД в группе 1 составила 9,4 %, в группе 3 – 10,5 %, группе 4 – 23,1 % и в группе 5 разница достигла 24,3 %. Увеличение толщины КИМ ОСА указывает на прогрессирующие структурно-функциональные изменения, связанные с замещением мышечно-эластического компонента сосудистой стенки на пролиферирующие грубоволокнистые соединительнотканные элементы, уменьшение микрососудистого аппарата и нарушение трофики оболочек сосудов.
Связь полиморфизма гена eNOS с показателями АД, КИМ и ЭТ-1 была изучена у ММЛ с признаками АГ. Результаты представлены в табл. 2. Получены статистически значимые различия при оценке показателей систолического АД (САД), диастолического АД (ДАД), КИМ ОСА и уровня ЭТ-1 в циркулирующей крови. Обследованные с генотипом ТС и СС имели более высокие значения САД и ДАД в сравнении с лицами с генотипом ТТ (р < 0,05). Толщина КИМ ОСА была больше у пациентов с генотипом ТС в сравнении с ММЛ с генотипом ТТ (р < 0,05). Еще большая разница получена в значениях КИМ ОСА у обследуемых с генотипами ТТ и СС (р < 0,01).
Таблица 1
Аллельный полиморфизм Т-786С промотора гена eNOS у ММЛ и в группе контроля
|
Генотипы |
ММЛ с АГ (N = 61) |
НАД (N = 50) |
р (F) |
OR |
±5 % |
χ2 |
df |
p (χ2) |
||
|
n |
% |
n |
% |
|||||||
|
ТТ |
10 |
16,4 |
24 |
48 |
0,0001 |
0,212 |
0,088–0,51 |
18,608 |
2 |
0,0001 |
|
ТС |
31 |
50,8 |
23 |
46 |
0,133 |
1,213 |
0,573–2,566 |
|||
|
СС |
20 |
32,8 |
3 |
6 |
0,0001 |
7,642 |
2,117–27,59 |
|||
|
Т |
51 |
41,8 |
71 |
71 |
0,0001 |
0,293 |
0,167–0,515 |
18,924 |
1 |
0,00001 |
|
С |
71 |
58,2 |
29 |
29 |
0,0001 |
3,408 |
1,943–5,98 |
|||
Таблица 2
Сравнительная характеристика значений АД, структурно-функциональных показателей эндотелия у лиц с признаками артериальной гипертензии в зависимости от полиморфизма Т-786С промотора гена eNOS
|
ТТ |
ТС |
СС |
|
|
10 |
31 |
20 |
|
|
САД (мм рт. ст.) |
140,3 (135,6–143,7) |
145,3 (141,7–148,8)* |
153,8 (146,5–162,1)# |
|
ДАД (мм рт. ст.) |
83,4 (77,2–89,9) |
90,6 (85,2–94,4) * |
97,5 (93,2–101,7)#° |
|
КИМ ОСА (мм) |
0,62 (0,55–0,70) |
0,77 (0,70–0,84) * |
0,87 (0,79–0,94) # |
|
ЭТ-1 (пмоль/л) |
13,49 (9,85–16,34) |
9,43 (6,41–11,85) |
52,54 (17,06–87,09)#° |
Примечания: р < 0,05: ТТ – ТС – *; ТТ – СС – #; ТС – СС –°.
Интересной оказалась выявленная зависимость распределения уровня ЭТ-1 в группах ММЛ с признаками АГ с разными генотипами. В группе ММЛ с генотипом ТС содержание ЭТ-1 снизилось по сравнению с исследуемой группой с генотипом ТТ. Вероятно, имела место активация eNOS с гиперпродукцией NO и усилением реакции с вазоактивным действием NO. Можно предположить, что наличие протективного аллеля Т у пациентов с гетерозиготным вариантом генотипа сопровождается увеличением каталитической активности eNOS и синтеза NO, что опосредованно ведет к подавлению и уменьшению синтеза ЭТ-1. В группе ММЛ с генотипом С отмечалось значительное увеличение содержания ЭТ-1, что указывало на снижение активности eNOS и выработки NO. Носители СС генотипа eNOS имели более высокие показатели АД и толщины КИМ. Угнетение активности синтеза NO сопровождается увеличением продукции ЭТ-1, который обладает широким спектром патологических эффектов, в частности вызывает пролиферацию фибробластов, избыточную продукцию внеклеточного матрикса.
Заключение
Таким образом, воздействие ХС способствует экспрессии генотипов СС и ТС полиморфного варианта Т-786С промотора гена eNOS у ММЛ, что приводит к изменению секреции вазоактивных веществ эндотелием сосудов в сторону увеличения продукции вазоконстрикторов (ЭТ-1) и снижению образования основного вазодилататора NO. Это способствует формированию АГ, процессов ЭД, активирует процессы ремоделирования сосудистой стенки с увеличением размеров КИМ ОСА, что определяет повышенный риск возникновения атеросклероза и сосудисто-мозговых событий. Внедрение данных клинико-генетических исследований в клиническую практику позволит специалистам различного профиля своевременно формировать группы повышенного риска возникновения АГ, разрабатывать и индивидуализировать мероприятия по превентивной коррекции возможного развития АГ.
В перспективе генетическое тестирование, в частности полиморфного варианта 786С промотора гена eNOS, может быть использовано при отборе в профессии, связанные с воздействием ХС, с целью выявления лиц с высоким риском развития АГ.
Рецензенты:
Туровская Т.В., д.м.н., профессор кафедры внутренней медицины № 1 Донецкого национального медицинского университета им. М. Горького, г. Донецк;
Багрий А.Э., д.м.н., профессор кафедры внутренних болезней, общей практики семейной медицины УНИПО Донецкого национального медицинского университета им. М. Горького, г. Донецк.
Работа поступила в редакцию 29.12.2014.
Библиографическая ссылка
Луцкий И.С., Зяблицев С.В., Луцкий Е.И., Кишеня М.С., Чернобривцев П.А. КЛИНИКО-ГЕНЕТИЧЕСКИЕ АСПЕКТЫ ФОРМИРОВАНИЯ АРТЕРИАЛЬНОЙ ГИПЕРТЕНЗИИ В УСЛОВИЯХ ДЕЙСТВИЯХ ХРОНИЧЕСКОГО СТРЕССА // Фундаментальные исследования. 2014. № 10-9. С. 1753-1758;URL: https://fundamental-research.ru/en/article/view?id=36507 (дата обращения: 12.07.2026).