


Scientific journal
Fundamental research
ISSN 1812-7339
"Перечень" ВАК
ИФ РИНЦ = 1,798
OXIDATIVE STRESS AND GLUTAMATERGIC EXCITOTOXICITY IN THE DEVELOPMENT OF DRUG-INDUCED TARDIVE DYSKINESIA (REVIEW)

Шизофрения является тяжелым психическим расстройством, которое выражается в нарушении психических функций, связана со структурными и поведенческими отклонениями и имеет неблагоприятный исход вне зависимости от применяемых методов лечения [7, 10, 11, 43, 21]. Основной группой психофармакологических средств, применяемых для лечения шизофрении, являются антипсихотические препараты (нейролептики), которые наряду с клиническими эффектами зачастую обладают широким спектром побочных эффектов [9]. Экстрапирамидные расстройства (паркинсонизм, дистония, тремор и т.д.) существенно снижают качество жизни больного. Среди них можно выделить такой вид гиперкинеза, как поздняя или тардивная дискинезия (ТД), возникающая у 20–30 % больных при длительном применении препаратов [8, 6]. Проявления дискинезии могут включать непреднамеренные движения орально-лицевой области, хореоатетоз конечностей (комбинация быстрых порывистых и медленных судорожных движений) и стереотипные ритмичные движения туловища [31]. Наиболее распространенным видом ТД является орофациальная дискинезия, которая наблюдается примерно в 80 % случаев гиперкинезов [23]. Большая часть дискинезий вызывается антипсихотиками первого поколения, но встречаются случаи, спровоцированные атипичными нейролептиками [24]. Несмотря на то, что в последние годы широко применяются атипичные антипсихотики для лечения шизофрении, классические нейролептики по-прежнему востребованы и распространены, так как у некоторых препаратов второго поколения наряду с аналогичным антипсихотическим эффектом имеются побочные проявления, не менее тяжелые, чем при применении традиционных нейролептиков [34]. Кроме того, фармакоэкономическая предпочтительность является немаловажным фактором в связи с более низкой стоимостью типичных антипсихотиков по сравнению с препаратами второго поколения [23].
Патогенез тардивной дискинезии до настоящего времени до конца не ясен [17, 35, 46]. Наследственный характер ТД предполагает сильную генетическую основу, в то же время такие факторы, как возраст, женский пол, диагноз сопутствующего аффективного расстройства и органическая дисфункция головного мозга, были также определены в качестве факторов риска [36]. Наиболее распространенными гипотезами возникновения тардивной дискинезии являются гипотезы гиперчувствительности дофаминовых рецепторов, серотонинергической дисфункции, ГАМКергической недостаточности и активации окислительного стресса [18]. Появление гиперкинеза при применении антипсихотиков, блокирующих D2-рецепторы, позволяет говорить об угнетении дофаминергической передачи. Помимо того, в ответ на блокаду рецепторов компенсаторно усиливается синтез и высвобождение дофамина, который активирует незаблокированные D1- или гиперчувствительные D2-рецепторы.
В связи с развитием дисбаланса в нейротрансмиттерной системе при шизофрении активируются процессы окислительного стресса [44, 33, 12, 47, 28]. Антипсихотики, благодаря своей липофильности, способны встраиваться в клеточные мембраны и нарушать метаболизм нейронов. В последние годы показано, что окислительный стресс и снижение антиоксидантной защиты способствуют гибели нейронов [33] и могут быть ассоциированы с развитием поздней дискинезии [13, 1]. У больных с тардивной дискинезией показано достоверное снижение количества восстановленного глутатиона в сыворотке крови и индекса отношения количества восстановленного глутатиона к окисленному глутатиону, что может быть использовано в качестве дополнительных параклинических критериев для прогноза риска развития побочных двигательных расстройств [3, 14].
Теория нейротоксичности предлагает объяснение многим клиническим особенностям ТД. Окислительный стресс со временем усиливает процессы деструкции, что выдвигает потенциальное объяснение того факта, что риск возникновения поздней дискинезии увеличивается с возрастом [44].
Важная роль в патогенезе тардивной дискинезии принадлежит глутаматергической системе [29]. Блокада дофаминовых рецепторов, регулирующих активность глутаматергических кортикостриарных терминалей, усиливает высвобождение глутамата, который оказывает эксайтотоксическое действие на ГАМК-ергические нейроны. Пресинаптические D2 рецепторы ингибируют высвобождение глутамата из нейронов головного мозга [29]. Таким образом, нейролептическая блокада этих рецепторов увеличивает синаптическое высвобождение аспартата и глутамата в стриатуме [18]. Как известно, стойкая активация ионотропных глутаматергических рецепторов является причиной нейрональной дегенерации. Окислительное повреждение опосредуется замедленной дегенерацией нейронов вследствие активации глутаматных NMDA- и не NMDA-рецепторов. Свободные радикалы могут повреждать клеточные белки, мембраны и ДНК, в зависимости от их происхождения, что в итоге приводит к гибели клеток [44]. Важным аспектом взаимосвязи замедленной глутамат-индуцированной дегенерации и окислительного стресса является то, что он обеспечивает механизм, посредством которого длительная медленная активация глутаматного рецептора может привести к кумуляции повреждающего эффекта, в конечном счете заканчивающегося дегенерацией нейронов (рисунок).
Эта форма нейродегенерации демонстрирует многие характеристики, свойственные апоптозу. Mitchell et al. [38] отмечают, что элиминация дофаминергических афферентов стриатума приводит к апоптозу нейронов полосатого тела. Кроме того, высокий уровень свободных радикалов может ингибировать пресинаптическое поглощение глутамата, инактивировать антиоксидантные ферменты и нарушить перенос электронов в митохондриальной дыхательной цепи. Свободные радикалы стимулируют образование NO-синтазы и, взаимодействуя с супероксидом, образуют высокореакционный радикал пероксинитрит [45], что приводит к повышенной генерации активных форм кислорода и внутринейронных возбуждающих аминокислот. Эти вторичные взаимодействия могут создавать замкнутый круг, активируя глутамат, опосредующий окислительное повреждение в стриатуме (рисунок).
Непосредственно сами антипсихотические препараты также могут обладать нейротоксическим действием: например, галоперидол в эксперименте in vitro вызывает шестикратное увеличение продукции активных форм кислорода в митохондриях [18]. Некоторые промежуточные продукты метаболизма антипсихотиков первого поколения обладают цитостатическим эффектом и потенциально связаны с лекарственно-индуцированными экстрапирамидными расстройствами.
В эксперименте на крысах было обнаружено, что длительное применение галоперидола уменьшает активность антиоксидантных ферментов супероксиддисмутазы и каталазы в мозге животного с соответствующим увеличением продуктов перекисного окисления липидов [40]. Эти исследования показывают, что галоперидол вызывает усиление окислительного стресса. Также было показано, что антиоксиданты (такие как мелатонин, кверцетин и AD4) уменьшают выраженность непреднамеренных движений жевательных мышц крыс на модели искусственно вызванной галоперидолом тардивной дискинезии [41].
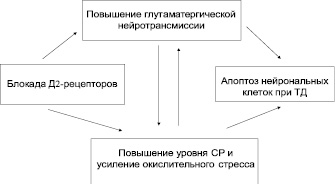
Гипотетическая модель нейролептик-индуцированного окислительного стресса и глутаматергической нейротрансмиссии при тардивной дискинезии
Сильным окислителем является оксид азота, который в случае избыточного синтеза приводит к повреждению нервной ткани. В случае искусственно вызванной ТД на моделях животных было обнаружено, что ингибитор NO-синтазы снижает выраженность непреднамеренных движений жевательных мышц у крыс. L-аргинин, являясь предшественником оксида азота, обращает этот эффект, что указывает на причастность NO к развитию нейролептик-индуцированной тардивной дискинезии [41]. Наблюдалось уменьшение экстрапирамидной симптоматики, вызванной хроническим введением галоперидола, при понижении уровня циклического гуанозинмонофосфата (цГМФ) в стриатуме, так же, как и концентрации оксида азота в сыворотке крови. NO является медиатором в случае NMDA-индуцированной нейротоксичности, которая может иметь значение в патофизиологии тардивной дискинезии. Оксид азота играет значительную роль и в регулировании концентрации дофамина, посредством как постсинаптических, так и пресинаптических механизмов [41]. Это примечательно в связи со значимостью теории нейротоксичности в развитии ТД, основу которой составляет гиперчувствительность дофаминергических рецепторов.
Возможные механизмы проокислительного/антиоксидантного дисбаланса в ЦНС включают в себя нарушения неферментативного и ферментативного метаболизма дофамина, нарушения в митохондриальной цепи переноса электронов и последствия применения антипсихотической терапии [20].
Известно, что генетические особенности являются причиной от 20 до 95 % всех неблагоприятных реакций организма человека на лекарственные соединения. Не все пациенты, получающие длительную антипсихотическую терапию, проявляют признаки экстрапирамидных расстройств, только у трети больных диагностируют дискинезию, что свидетельствует о наличии индивидуальных особенностей в развитии ТД [23]. Литературные данные убедительно демонстрируют ассоциацию ТД с генетическими факторами [36]. Ряд исследований предполагает возникновение индивидуальной вариации для развития дискинезии вследствие взаимодействия генов как друг с другом, так и с окружающей средой. Основными направлениями фармакогенетических исследований лекарственно-индуцированной тардивной дискинезии являются: выявление ассоциаций полиморфизмов генов системы метаболизма ксенобиотиков (гены первой и второй фаз метаболизма антипсихотиков), генов нейромедиаторных рецепторов и генов окислительного стресса [15, 16, 2, 42, 29].
Достаточно большое количество фармакогенетических исследований направлено на поиск ассоциаций полиморфизмов генов, кодирующих основные ферменты антиоксидантной защиты. Нарушения синтеза антиоксидантов или активности антиоксидантных ферментов, вследствие носительства определенного генотипа, могут увеличить восприимчивость к окислительному стрессу, что может привести к нарушению функций нейромедиаторов в головном мозге [21, 45], как центрального, так и периферического происхождения. Имеющиеся литературные данные подтверждают, что периферические показатели нейромедиаторов отражают их состояние в ЦНС [22]. Тяжесть реализации экстрапирамидной симптоматики может быть потенциально связана с биомаркерами окислительного стресса [45, 48].
Убедительно продемонстрированы ассоциации полиморфизмов гена, кодирующего марганецзависимую супероксиддисмутазу (MnSOD), митохондриального энзима, вовлеченного в оксидативный метаболизм [19, 46]. Замена аланина на валин (Ala9Val) приводит к снижению функциональной активности MnSOD в митохондриях. Как сообщается, функциональный однонуклеотидный полиморфизм (SNP) (Ala-9Val) в гене MnSOD связан с развитием тардивной дискинезии в японской, польской и африканской популяции [26] и российской популяции [15]. Zhang et al. [48] обнаружил достоверную положительную корреляцию между показателями шкалы непреднамеренных двигательных расстройств AIMS, по которой оценивается тяжесть ТД, и активностью MnSOD.
С точки зрения фармакогенетики важное значение также имеют полиморфизмы генов, контролирующих синтез и работу ферментов биотрансформации лекарственных средств, например, глутатион-S-трансфераза (GST), которая выполняет детоксикационную и антиоксидантную функции [3, 15]. GST осуществляет конъюгацию сульфгидрильной (SH) группы глутатиона с ксенобиотиками или их метаболитами, образовавшимися в первой фазе биотрансформации. Данная реакция играет ведущую роль в защите клеток от свободных радикалов [32]. Синтез глутатионтрансфераз контролируется различными генами, в которых выявлены однонуклеотидные замены, оказывающие существенное влияние на их функции. Одним из важнейших свойств системы метаболизма является индукция – активация транскрипции гена в присутствии субстрата. Тканеспецифичная экспрессия различных изоформ метаболизма ксенобиотиков определяет ее адаптацию к структурно-функциональной организации той или иной системы организма. При изучении аллельных вариантов гена GSTP1 (rs1695) было выявлено, что генотип AG и аллель G гена глутатион-S-трансферазы (313A > G) имеют протективное значение, снижая риск развития тардивной дискинезии у больных шизофренией, получающих антипсихотики [2]. Эти знания о полиморфизме генов ферментов метаболизма антипсихотиков позволяют лучше понять процессы биотрансформации ксенобиотиков и, как следствие, могут внести определенный вклад в изучение их побочных эффектов, в том числе и тардивной дискинезии.
Таким образом, патофизиология и патогенез экстрапирамидных расстройств, возникающих на фоне приема нейролептических препаратов, окончательно не выяснены. Тардивная дискинезия является мультифакториальным расстройством, в развитии которого важную роль играют как процессы окислительного стресса, так и полиморфизмы генов антиоксидантных ферментов. Дальнейшие биохимические и фармакогенетические исследования в отношении механизмов развития тардивной дискинезии имеют перспективу в рамках концепции персонализированной терапии и будут направлены на разработку молекулярно-диагностических маркеров для прогнозирования риска развития тардивной дискинезии, что позволит применять индивидуальную терапевтическую тактику.
Обзор написан в рамках выполнения грантов РФФИ № 14-04-31876 «Эксайтотоксичность и деструктивные процессы в патогенезе лекарственно-индуцированных двигательных расстройств у больных шизофренией» и № 12-04-33072 «Патогенез двигательных нарушений у больных эндогенными психическими расстройствами на фоне антипсихотической терапии: роль фармакогенетических факторов».
Рецензенты:
Семке А.В., д.м.н., профессор кафедры психиатрии, наркологии и психотерапии, ГБОУ ВПО «Сибирский государственный медицинский университет» Министерства здравоохранения и социального развития Российской Федерации, г. Томск;
Невидимова Т.И., д.м.н., профессор кафедры нормальной физиологии, ГБОУ ВПО «Сибирский государственный медицинский университет» Министерства здравоохранения и социального развития Российской Федерации, г. Томск.
Работа поступила в редакцию 02.12.2014.
Библиографическая ссылка
Бойко А.С. ОКИСЛИТЕЛЬНЫЙ СТРЕСС И ГЛУТАМАТЕРГИЧЕСКАЯ ЭКСАЙТОТОКСИЧНОСТЬ В РАЗВИТИИ ЛЕКАРСТВЕННО-ИНДУЦИРОВАННОЙ ТАРДИВНОЙ ДИСКИНЕЗИИ (ОБЗОР ЛИТЕРАТУРЫ) // Фундаментальные исследования. 2014. № 10-6. С. 1220-1226;URL: https://fundamental-research.ru/en/article/view?id=36020 (дата обращения: 30.06.2026).