


Scientific journal
Fundamental research
ISSN 1812-7339
"Перечень" ВАК
ИФ РИНЦ = 1,798
PARTIAL DELETIONS OF FOXK1 IN CHILDREN WITH AUTISM: IDENTIFICATION OF A NEW CANDIDATE GENE OF AUTISTIC SPECTRUM DISORDERS BY POSTGENOMIC TECHNOLOGIES

Многие работы в области биологической психиатрии и психиатрической генетики последних лет демонстрируют связь между аутизмом и геномными вариациями. У детей с аутистическими расстройствами выявляются регулярные структурные хромосомные аберрации, включая интерстициальные микроделеции/микродупликации, и вариации числа копий последовательностей ДНК (CNV), затрагивающие гены, многие из которых рассматриваются в качестве кандидатов для этого заболевания [3–6, 11]. С внедрением методов, позволяющих сканировать геном с высоким разрешением (в частности, молекулярное кариотипирование), количество позитивных ассоциаций между геномной патологией и аутизмом в значительной степени увеличилось [2, 6, 10]. Помимо этого, применение технологий изучения межклеточных вариаций генома также демонстрирует большое число случаев (до 16 %) аутистических расстройств, связанных с соматическим мозаицизмом (наличие нескольких популяций клеток в организме, отличающихся друг от друга по геномному составу) [7, 8, 16]. Более того, вариабельность участков генома, содержащих последовательности ДНК, которые до сих пор остаются фактически неизвестными (гетерохроматиновые участки хромосом), также считается одним из факторов риска развития аутизма [1, 15]. Создание унифицированного каскада патогенетических процессов, включающего весь широкий спектр вариабельности генома при данных формах нарушения психики, является одним из приоритетов в современной психиатрической генетики, в рамках основной парадигмы которой поиск генов-кандидатов аутизма считается основополагающим. Наличие соответствующих данных также позволяет определять молекулярные механизмы заболевания с целью последующей разработки научно обоснованной терапии [5].
Целью настоящей работы явился поиск генов-кандидатов в группе 64 детей с аутизмом с помощью постгеномных технологий: молекулярное кариотипирование или высокоразрешающее сканирование генома с разрешением не менее 1 тыс. пн и биоинформатический анализ, включающий в себя оценку патогенности вариаций генома с помощью геномных, эпигеномных и протеомных баз данных, а также моделирования белковых взаимодействий (интерактомный анализ).
Материалы и методы исследования
Полногеномное сканирование было использовано для анализа 64-х образцов ДНК периферической крови детей с клиническим диагнозом «аутистические расстройства», входящих в российскую когорту детей с аутизмом, описанную ранее [1, 2, 5, 10, 15, 16]. Молекулярное кариотипирование проводилось с использованием высокоразрешающих чипов Affymetrix (CytoScan HD) для SNP (single nucleotide polymorphism)/олигонуклеотидной сравнительной геномной гибридизации (разрешение 1000 пн и более) в соответствии с протоколами, представленными ранее [2, 10]. Биоинформатический анализ выявленных перестроек включал в себя оценку патогенности с помощью геномных, эпигеномных/транскриптомных, протеомных и метаболических баз данных. Оригинальная биоинформатическая технология подробно описана в предыдущих работах [9, 10]. Интерактомная цепочка для анализа взаимодействий белка, кодируемым геном FOXK1, была получена с использованием программы Cytoscape 2.8.3 [12].
Результаты исследования и их обсуждение
В ходе анализа генома 64 пациентов у двух (3,1 %) были обнаружены CNV в виде делеций последовательности ДНК гена FOXK1. В первом случае делеция затронула семь экзонов гена FOXK1 (с третьего по девятый) и гены KIAA0415 и RADIL. Биоинформатический анализ этих двух генов показал, что вероятность их ассоциации с аутистическими расстройствами крайне мала. Во втором случае делеция затронула первый экзон гена FOXK1 (рис. 1).
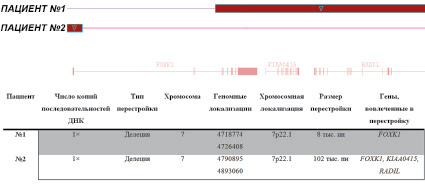
Рис. 1. Схематическое изображение делеций, затронувших ген FOXK1, которые были выявлены у двух пациентов с помощью полногеномного сканирования, с обозначением геномной и хромосомной локализации, размера перестройки и других генов, вовлеченных в перестройку
Примечательно, что вариации последовательностей ДНК гена FOXK1 ранее не были ассоциированы с какими-либо фенотипическими проявлениями. Ранее было лишь показано, что ген FOXK1 обладает повышенной экспрессией в нервных клетках у млекопитающих [13] и вовлечен в геномную цепочку регуляции пролиферации клеток [14]. С другой стороны, биоинформатический анализ показывает, что данный ген, скорее всего, кодирует белок-регуляции транскрипции. Интерактомный анализ (изучение межбелковых взаимодействий продукта транскрипции гена-мишени) (рис. 2) свидетельствует о том, что нарушение функционирования FOXK1 связано с изменениями в следующих внутриклеточных процессах: регуляция митотического деления клеток, апоптоза и экспрессии генов (за счет нарушения взаимодействия с YWHAB); ремоделирование хроматина (за счет нарушения взаимодействия с BAP1); внутриутробное развитие (за счет нарушения взаимодействия с AMOT); метаболизм ионов металлов (за счет нарушения взаимодействия с FYCO1).
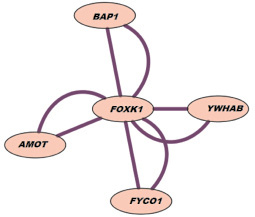
Рис. 2. Результаты интерактомного анализа, демонстрирующего взаимодействие белка, кодируемого геном FOXK1 с помощью программы Cytoscape 2.8.3 [12]
Примечательно, что нарушения регуляции митоза и/или апоптоза рассматриваются как факторы риска при аутизме, являясь причиной хромосомного мозаицизма и нестабильности, наблюдаемых при этом заболевании [4–7, 15, 16], а изменения таких эпигенетических феноменов, как регуляция экспрессии генов и ремоделирование хроматина неоднократно ассоциировались с различными формами аутистических расстройств [1, 5, 6, 11]. Совокупность полученных биоинформатических данных позволяет сделать вывод о том, что мутации в гене FOXK1 могут приводить к нарушениям психики, а вывод о том, что он является геном-кандидатом аутизма, является обоснованным.
Заключение
Анализ генома с целью выявления генов-кандидатов аутистических расстройств и патогенетических процессов, характерных для этого заболевания, является неотъемлемой частью современных исследований в области психиатрической генетики и биологической психиатрии [3–8, 11]. Однако, несмотря на повышенный интерес к генетике аутизма, существует ряд проблем при выявлении молекулярных основ подобных форм нарушения психики, связанных преимущественно с интерпретацией обнаруженных геномных вариаций. Преодоление подобных сложностей, как было показано ранее [9, 10], возможно только при использовании биоинформатических технологий в дополнение к методам высокоразрешающего сканирования генома. Применяя комплекс постгеномных технологий, сочетающий молекулярное кариотипирование и биоинформатический анализ, был обнаружен новый ген-кандидат аутизма. В связи с этим, принимая во внимание потенциал сканирования генома и оригинальных технологий in silico, использованных в настоящей работе, можно с уверенностью утверждать, что описываемый подход к поиску генетических изменений, приводящих к аутизму, является эффективным.
Исследование выполнено за счёт гранта Российского Научного Фонда (проект № 14-35-00060).
Работа поступила в редакцию 10.10.2014.
Библиографическая ссылка
Юров И.Ю., Ворсанова С.Г., Васин К.С., Коростелев С.А., Юров Ю.Б. ЧАСТИЧНЫЕ ДЕЛЕЦИИ ГЕНА FOXK1 У ДЕТЕЙ С АУТИЗМОМ: ОПРЕЛЕНИЕ НОВОГО ГЕНА-КАНДИДАТА АУТИСТИЧЕСКИХ РАССТРОЙСТВ С ПОМОЩЬЮ ПОСТГЕНОМНЫХ ТЕХНОЛОГИЙ // Фундаментальные исследования. 2014. № 10-4. С. 767-770;URL: https://fundamental-research.ru/en/article/view?id=35620 (дата обращения: 01.07.2026).