


Scientific journal
Fundamental research
ISSN 1812-7339
"Перечень" ВАК
ИФ РИНЦ = 1,798
IMMUNOMORPHOLOGICAL FEATURES OF POSTINFECTION PULMONARY FIBROSIS IN HUMANS INFECTED WITH INFLUENZA A/H1N1 VIRUS

С 2009 года во всем мире регистрировали случаи инфицирования населения новым штаммом вируса гриппа A/H1N1, при этом тяжелое течение и летальные исходы наблюдали не только у людей из групп риска, но и у здорового населения [6]. В связи с массовой заболеваемостью населения новым штаммом вируса гриппа А, ВОЗ объявила о 6-й фазе угрозы пандемии гриппа [15]. При изучении нового штамма вируса гриппа А оказалось, что он является трижды реассортантным, что в определенной степени объясняет наличие некоторого уровня антител против данного штамма у лиц старше 60 лет и их полное отсутствие у людей репродуктивного возраста [5]. Одним из осложнений гриппа А является прогрессирующий фиброз легких, который ухудшает качество жизни больных, а также приводит к последующей инвалидизации и смерти [13].
В общем виде развитие фиброза легких вне связи с конкретным этиологическим фактором происходит по следующей схеме:
1) повреждения базальной мембраны эпителия / эндотелиального барьера;
2) секреция TGF β1 – основного профибротического цитокина;
3) избыточная экспрессия гиалуронана;
4) рекруция воспалительных клеток;
5) образование активных форм кислорода;
6) активация коллаген-продуцирующих клеток;
7) активация внеклеточного матрикса фибробластами.
Фиброзная ткань рассматривается в качестве окончательного следствия, для которого пока не существует успешной терапии [9].
Помимо фибробластов важную роль в процессах фиброгенеза играют и макрофаги, одни из основных цитокин-продуцирующих клеток организма. Многие авторы в основе патогенеза раннего фиброзирования при инфицировании или стимуляции макрофагов чужеродным антигеном отмечали неконтролируемый иммунный ответ, связанный с гиперцитокинемией [10, 12]. В этой связи, можно предполагать, что высокопатогенные вирусы, в том числе вирусы гриппа А, индуцируют фиброз как за счет прямых механизмов, связанных с внутриклеточной персистенцией и репликацией самих возбудителей [14], так и путем цитокин-опосредованной регуляции [2, 8].
Поскольку ежегодные эпидемии гриппа связаны прежде всего с реассортантными штаммами вируса гриппа А, представляется актуальным дальнейшее изучение особенностей его пато- и морфогенеза.
Материалы и методы исследования
По протоколам аутопсий было проанализировано 6 случаев смерти пациентов в ГБУЗ НСО «Городская инфекционная клиническая больница №1» (г. Новосибирск), у которых прижизненно было проведено вирусологическое исследование, выявившее наличие антител к вирусу гриппа А (H1N1) методом полимеразной цепной реакции в назофарингиальных образцах. Среди умерших были 5 мужчин, средний возраст которых составил 37,2 ± 4,03 года, и 1 женщина в возрасте 26 лет. Длительность заболевания составляла 14 ± 2,96 суток (от 6 до 22 суток). Длительность пребывания в стационаре 8,5 ± 2,27 койко-дня (от 1 до 18 койко-дней). Смерть больных наступила в период от 10 до 24 суток от начала заболевания. По времени смерти пациенты были условно разделены на 2 группы: группа «ранней» гибели (10–12 сутки от начала заболевания) и группа «поздней» гибели (22–24 сутки). В качестве контроля были взяты аутопсийные случаи 10 больных в возрасте от 22 до 38 лет, умерших от заболеваний не связанных с патологией легких и не имеющих в анамнезе хронических неспецифических заболеваний легких.
По протоколам аутопсий определяли наличие сочетанной и сопутствующей патологии и макроскопические изменения внутренних органов. Парафиновые срезы окрашивали гематоксилином и эозином, пикрофуксином по методу ван Гизон. Иммуногистохимическое (ИГХ) исследование проводили непрямым пероксидазным методом с использованием первичных антител: антиген вируса гриппа A/H1N1, меченного FITC (Influenza A) («Abcam»), маркер фибробластов («Diagnostic BioSystems»), CD68 («Diagnostic BioSystems»), индуцибельная NO–синтаза (iNOS) («Novocastra»), TGFR1-3 («Abcam»), TNF-α («Diagnostic BioSystems»).
Для проведения ИГХ-исследования парафиновые срезы легких подвергали депарафинизации, регидратации, демаскировке антигенов в микроволновой печи мощностью 700 Вт. Экспозицию с первичными антителами проводили согласно инструкциям производителей. Затем срезы инкубировали с HRP-конъюгатом и вторичными антителами (система детекции «Spring Bioscience»), далее обрабатывали DAB-субстратом и дополнительно окрашивали гематоксилином Майера. Визуализацию проводили на микроскопе AxioImager A1 с фотокамерой AxioCam MRc (Carl Zeiss).
Топологию вируса определяли методом иммуннофлуоресценции – непрямого анализа с применением первичных антител на антиген вируса Influenza A с использованием конфокального лазерно-сканирующего микроскопа LSM 710 (Carl Zeiss).
Для морфометрии использовали закрытую тест-систему из 100 точек площадью 3,52х104 мкм2 и инструментов программы AxioVision (rel. 4.8.2). Определяли объемную (Vv) плотность соединительной ткани (коллагеновых волокон), численную плотность (Nai) фибробластов, численную плотность (Nai) клеток, экспрессирующих антиген вируса гриппа А, численную плотность (Nai) макрофагов, экспрессирующих iNOS, TNF-α и численную плотность (Nai) фибробластов, экспрессирующих TGFR1-3. Кроме того, производили расчет «фибропластической активности» по соотношению объемной плотности коллагеновых волокон, приходящихся на один фибробласт [1], в закрытой тестовой системе.
Средние величины исследованных параметров определяли с помощью пакета анализа данных программы STATISTICA v.6. Для выявления степени вероятности достоверности различий сравниваемых средних величин применяли t-критерий Стьюдента. Достоверными считали различия при р < 0,05.
Результаты исследования и их обсуждение
На аутопсии у всех больных были выявлены признаки трахеобронхита, представленного различными морфологическими формами от катарально-эрозивного до гнойно-некротического. Кроме того, во всех 6 случаях была обнаружена двусторонняя пневмония: в 4 случаях вирусно-бактериальная и в 2 случаях вирусная. Осложнения были представлены геморрагическим отеком легких, а также в трех случаях отеком и набуханием головного мозга. Также в качестве осложнений встречались двусторонний гидроторакс, тромбоз подключичной и плечеголовной вен и острый абсцесс легкого.
Из представленных в табл. 1 данных видно, что у 80 % умерших имелись сопутствующие заболевания. В подавляющем большинстве – это ожирение разной степени выраженности (65 %), в половине случаев встречалась артериальная гипертензия, а также стеатоз печени (30 %). В одном случае в качестве сопутствующего заболевания выступали гемофилия А с хроническим гепатитом С, у одного больного в анамнезе сахарный диабет II типа.
Таблица 1
Сопутствующие и сочетанные заболевания
|
№ п/п |
Возраст, пол |
Заболевания |
|
1 |
38 л., муж. |
Гемофилия А. Хронический гепатит С умеренной степени активности, IV стадия фиброза |
|
2 |
30 л., муж. |
Ожирение III степени. Стеатоз печени |
|
3 |
30 л., муж. |
- |
|
4 |
36 л., муж. |
Ожирение III степени. Стеатоз печени. Артериальная гипертензия II, гипертрофическая кардоимиопатия |
|
5 |
26 л., жен. |
Артериальная гипертензия II. Ожирение I-II степени. |
|
6 |
52 г., муж. |
Сахарный диабет II типа. Артериальная гипертензия. Ожирение III степени |
У всех больных, независимо от длительности заболевания был обнаружен вирусный антиген в отделяемом носоглотки. Также, методом иммунофлюоресценции была обнаружена экспрессия вирусного антигена в различных клетках легких. Антиген вируса гриппа А, экспрессировался преимущественно макрофагами и альвеолоцитами, также в небольшом количестве была обнаружена его экспрессия в эндотелиоцитах и единичных фибробластах. Общее количество клеток, экспрессирующих вирусный антиген, составляло от 33 до 75 клеток в тестовой площади, в зависимости от длительности заболевания. Следует отметить, что вирусный антиген определяли в клетках легких вплоть до 24 суток заболевания, что может быть свидетельством пролонгированной персистенции вируса гриппа A (H1N1). При этом имело место нарастание количества инфицированных клеток к 12 суткам с последующим снижением.
При микроскопическом исследовании легких во всех случаях, независимо от сопутствующей патологии, были обнаружены сходные морфологические изменения: диффузная лимфомакрофагальная инфильтрация с примесью нейтрофилов; интерстициальный и альвеолярный отек; кровоизлияния; полнокровие и тромбоз сосудов микроциркуляторного русла. Также отмечали десквамацию альвеолярного и бронхиального эпителия, плоскоклеточную метаплазию эпителия трахеи и бронхов. Кроме патоморфологических изменений, характерных для трахеобронхита и вирусной/вирусно-бактериальной пневмонии, у всех умерших пациентов было обнаружено избыточное разрастание соединительной ткани, по сравнению с умершими из группы контроля (рис. 1), преимущественно в перибронхиальных и периваскулярных зонах, а также в интерстиции. Объемная плотность соединительной ткани в легких пациентов составляла от 4,5 до 10,8 %, что превосходило контрольные значения от 2 до 5 раз.
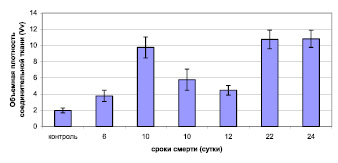
Рис. 1. Объемная плотность соединительной ткани в легких пациентов, инфицированных вирусом гриппа A/H1N1, и пациентов из группы контроля (аутопсийное исследование)
Поскольку в процессе фиброгенеза основным «источником» синтеза компонентов внеклеточного матрикса является фибробласт [9], можно полагать, что увеличение волокнистой соединительной ткани в легких прежде всего связано с увеличением количества фибробластов или с повышением их «фибропластической активности».
При иммуногистохимическом исследовании в легких больных, инфицированных вирусом гриппа A/H1N1, наблюдали увеличение числа фибробластов, максимальное количество которых отмечали у больного умершего на 6 сутки от начала заболевания. При этом «фибропластическая активность» была наибольшей у больного, смерть которого наступила на 22 сутки от начала болезни (табл. 2).
Таблица 2
Численная плотность фибробластов легких и их фибропластическая активность у пациентов, инфицированных вирусом гриппа A/H1N1, и пациентов из группы контроля (аутопсийное исследование)
|
Номер случая |
Длительность заболевания (сутки) |
Фибропластическая активность |
Nai фибробластов |
|
контроль |
– |
0,18 |
11,05 ± 0,45 |
|
1 |
6 |
0,12 |
32,61 ± 3,17 |
|
2 |
10 |
0,54 |
18,07 ± 1,54 |
|
3 |
10 |
0,23 |
25,15 ± 2,72 |
|
4 |
12 |
0,24 |
18,58 ± 1,65 |
|
5 |
22 |
0,62 |
17,31 ± 0,63 |
|
6 |
24 |
0,43 |
24,89 ± 2,06 |
Исходя из полученных данных, можно предполагать, что подобный феномен связан с тем, что в ранние сроки заболевания происходит активная пролиферация фибробластов в ответ на массивное повреждение такни легких и гипоксию, а также в ответ на персистенцию вируса в макрофагах, альвеолоцитах и фибробластах. По мере увеличения сроков заболевания происходит созревание и дифференцировка юных фибробластов в миофибробласты [4], часть из которых погибает, но при этом, у сохранившихся миофибробластов увеличивается их фибропластическая активность.
Как известно, TNF-α обладает способностью стимулировать не только пролиферацию фибробластов, но и синтез ими коллагена [11]. При иммуногистохимическом исследовании образцов легких всех умерших пациентов, независимо от срока заболевания, было обнаружено повышенное количество макрофагов с экспрессией TNF-α (рис. 2), минимальное количество которых наблюдали у пациента, умершего на 12 сутки заболевания, при этом максимально высокие значения наблюдали у больного, умершего на 22 стуки заболевания.
Согласно данным, полученным с помощью корреляционного анализа, проводимого между величинами численной плотности макрофагов, экспрессирующих TNF-α и «фибропластической активностью» фибробластов была выявлена положительная умеренная корреляционная связь (r = 0,521). Это свидетельствует о том, что по мере увеличения длительности заболевания происходит нарастание уровня TNF-α, который способствует повышению фибропластической активности фибробластов, а соответственно увеличению объемной плотности соединительной ткани.
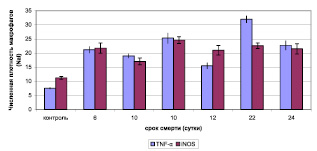
Рис. 2. Экспрессия TNF-α и iNOS макрофагами легких у пациентов, инфицированных вирусом гриппа A/H1N1, и пациентов из группы контроля (аутопсийное исследование)
Эндотоксемия и системное воспаление, способствующие повреждению тканей и продукции NO-синтаз фагоцитами, обусловливает избыточное накопление активных кислородных радикалов (ROS), таких как супероксид, перекись водорода и гидроксильные радикалы, тесно связанных с фиброзом [3], действующих как интрацеллюлярные вторичные посредники, оказывая профиброгенные эффекты. При проведении ИГХ-исследования в легких пациентов, инфицированных вирусом гриппа A/H1N1, регистрировали увеличение числа клеток различного гистогенеза, экспрессирующих iNOS, наибольшее количество которых составляли макрофаги (рис. 2). Кроме того, была выявлена положительная умеренная корреляционная связь (r = 0,418) между показателями экспрессии iNOS и количеством фибробластов, что, по всей видимости, говорит о повышении пролиферативной активности фибробластов под воздействием оксида азота.
Немаловажную роль в развитии фиброза при инфицировании вирусом гриппа A/H1N1 играет TGF-β и его рецептор. TGF-β регулирует свою продукцию макрофагами по аутокринному типу и стимулирует моноциты к секреции других факторов роста, участвующих в регенерации тканей, а в случае избыточной продукции, способствующие фиброзу [7]. У пациентов, инфицированных данным вирусом при ИГХ исследовании по сравнению с группой контроля, наблюдали увеличение количества фибробластов, экспрессирующих рецептор данного фактора роста (рис. 3). Наибольшее количество фибробластов, экспрессирующих TGFR наблюдали на 6 и 10 сутки болезни с уменьшением из числа в последующие сроки заболевания. При этом обращает на себя внимание тот факт, что количество фибробластов, экспрессирующих TGFR не имеет корреляционной зависимости с другими изучаемыми в данном исследовании параметрами, что говорит о его «независимости» от данных факторов. Вероятно, активация рецептора TGF происходит в ответ на гипоксию и повреждение клеток. Исходя из полученных данных следует, что повышенная экспрессия TGFR и последующее связывание его с TGF, вероятно, способствуют стимуляции пролиферации фибробластов и синтезу ими компонентов внеклеточного матрикса, приводящим к увеличению общего объема соединительной ткани в легких инфицированных вирусом гриппа А больных.
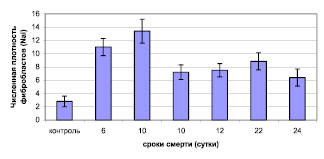
Рис. 3. Численная плотность фибробластов легких, экспрессирующих TGFR у пациентов, инфицированных вирусом гриппа A/H1N1, и пациентов из группы контроля (аутопсийное исследование)
Таким образом, развитие фиброза легких при гриппе A/H1N1 имеет многофакторную природу с участием различных механизмов. В основе же формирования и поддержания дисбаланса в системе ВКМ лежит персистенция вируса. Подобные механизмы требуют дальнейшего изучения.
Рецензенты:
Надеев А.П., д.м.н., профессор кафедры патологической анатомии, ГБОУ ВПО «Новосибирский государственный медицинский университет» Минздрава РФ, г. Новосибирск;
Сажина Т.В., д.м.н., профессор кафедры гистологии, эмбриологии и цитологии, ГБОУ ВПО «Новосибирский государственный медицинский университет» Минздрава РФ, г. Новосибирск.
Работа поступила в редакцию 05.08.2014.
Библиографическая ссылка
Аникина А.Г., Потапова О.В., Ковнер А.В., Черданцева Л.А., Шаркова Т.В., Шкурупий В.А., Иванов Г.Я. ИММУНОМОРФОЛОГИЧЕСКИЕ ОСОБЕННОСТИ ПОСТИНФЕКЦИОННОГО ПНЕВМОФИБРОЗА У ЧЕЛОВЕКА ПРИ ГРИППЕ A/H1N1 // Фундаментальные исследования. 2014. № 10-1. С. 18-23;URL: https://fundamental-research.ru/en/article/view?id=35205 (дата обращения: 10.08.2026).