


Scientific journal
Fundamental research
ISSN 1812-7339
"Перечень" ВАК
ИФ РИНЦ = 1,798
THE ANALYSIS OF HEREDITARY INFRINGEMENTS OF SYSTEM OF BLOOD IN THREE REGIONS OF AZERBAIJAN

Среди генетически обусловленных заболеваний системы крови важное место по значимости и распространенности занимают наследственные энзимопатии эритроцитов, вызванные дефицитом фермента глюкозо-6-фосфатдегидрогеназы (Г6ФД), который особенно характерен для стран Средиземноморья, некоторых стран Латинской Америки и Африки [13]. По сведениям ВОЗ, в мире, в том числе в СНГ, насчитывается около 100 млн. человек с недостаточностью Г6ФД [3; 4]. Ввиду расширения спектра производства и потребления в эндемичных регионах лекарственных препаратов окислительного действия и возникновения желтухи у больных новорожденных, неонатальные скрининг-программы по дефициту Г6ФД являются обязательными [9]. Наиболее распространенными моногенными расстройствами гемоглобинной системы во всем мире являются талассемии, среди которых β-талассемия составляет более 90% всех типов заболевания и эндемично для популяций Средиземноморского бассейна, Ближнего Востока, Азии и Африки. Растущая глобальная миграция привела к проникновению этого заболевания в США и северо-запад Европы, в виду чего скрининг на носительство β-талассемии входит в список обязательных профилактических программ во многих странах [7; 10; 11; 12]. Наиболее часто встречающимся наследственным геморрагическим диатезом коагуляционного генеза является гемофилия с частотой от 6,6 до 18 на 100.000 жителей мужского пола [1]. В виду того, что учитывается лишь обращаемость во время кровотечений, численность больных гемофилией в действительности оказывается гораздо больше [6]. Отмечая особую роль указанных заболеваний в инвалидизации детей, в том числе и в Азербайджане, результатом чего является длительное, сложное и дорогое лечение, нами проведены популяционно-генетические исследования среди населения Муганской, Ширванской и Западной зон Азербайджана с целью идентификации лиц, отягощенных энзимопенической анемией, β-талассемией и гемофилией, определения частоты распространения и типов мутаций заболеваний, что необходимо для дальнейшего проведения пренатальной диагностики в семьях с риском рождения больного ребенка и составления регистра по данным патологиям.
Материалы и методы
Материал собран в экспедиционных условиях в селах и в районных центрах Муганской, Ширванской и Западной зон Азербайджана в период с 2004 по 2007 гг. Для выявления больных с наследственной патологией крови использованы списки ВТЭК ЦРБ. В селах при подворовом обходе семей пробандов составлены родословные и путем генеалогического анализа дифференцированы случаи наследственных нарушений крови. В качестве материала для анализов использованы образцы крови, забор которой производили в микропробирки с антикоагулянтом. С помощью скрининг-программ выявлены случаи β-талассемии и недостаточности фермента Г6ФД среди учащихся 5-11-х классов [14]. В качестве экспресс-диагностики дефицита Г6ФД использован метод флюоресцирующих пятен Beutler E. (аппарат Hoefer MacroVue UV-25, Amersham Bioscience, USA) [2]. Для идентификации типа мутации β-талассемии на аппарате Thermal cycler Biocycler TC-S (Roche, USA) использован молекулярный метод высокотемпературной аллель-специфической амплификации, основанный по принципу метода полимеразно-цепной реакции [8; 17]. С целью тестирования изменений различных регионов β-глобинового гена (β-ГГ) использованы синтетические олигонуклеотидные контрольные праймеры - №№ 15, 16, 30, 31, и праймеры конкретных мутаций - 40 IVS-1-110 M (G-A), 41 IVS-1-110 N, 49 IVS-2-1 M (G-A), 77 IVS-2-1 N, 46 IVS-1-6 M (T-S), 54 Codon 8 M (-AA), 55 Codon 8 N. Для уточнения клинического диагноза больных наследственными гемоглобинопатиями использованы метод электрофореза гемоглобина на ацетат-целлюлозных пленках (аппарат EPS3501XL, USA) и аналитический метод изоэлектрофокусирования гемоглобинов в полиакриламидно-амфолиновых пластинках c рН 3,5-9,5 (аппарат Multiphor II, USA) [15; 16]. Частоты определены по общепринятой методике [5].
Результаты и обсуждение
В ходе проведенных исследований в 9-ти крупных районах трех зон Азербайджана выявлено 182 больных, среди которых у 103 детей установлена гемолитическая болезнь новорожденных (ГБН) - 56,60%, у 46 больных - β-талассемия с частотой 25,27%, у 33 больных - гемофилия с частотой 18,13% (рис.1).
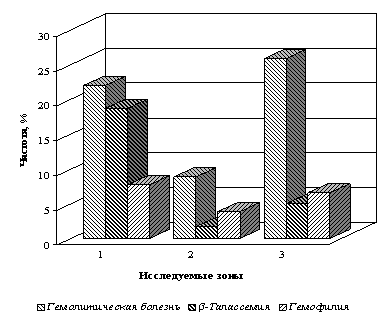
Рис.1. Наследственные болезни крови в трех зонах Азербайджана
1. Муганская зона; 2. Ширванская зона; 3. Западная зона
В данных популяциях идентифицировано 65 дефектных β-ГГ и пять типов мутаций β-талассемии: замена гуанина на аденин в 110-ой позиции I интрона b-ГГ с фенотипом b+-IVS-1-110(G-A); делеция двух нуклеотидов аденин в 8-ом кодоне I экзона β-ГГ - β0-codon 8(-АА); замена гуанина на аденин в 1-й позиции II интрона β-ГГ с фенотипом β0-талассемии - β0-IVS-2-1(G-A); замена гуанина на цитозин в 5-й позиции I интрона b-ГГ - b+-IVS-1-5(G-S); замена тимина на цитозин в 6-й позиции I интрона β-ГГ - β+-IVS-1-6(T-S). Результатом IVS-1-110(G-А), IVS-2-1(G-А), IVS-1-6(T-S), IVS-1-5(G-S) дефектов является нарушение этапа сплайсинга биосинтеза, результатом микроделеции codon 8(-АА) - нонсенс-мутация, нарушающая транскрипцию (табл. 1).
Таблица 1. Частота мутаций β-глобинового гена в трех зонах республики
|
Тип мутаций |
Частота генов, % |
||
|
Зоны |
|||
|
Муганская |
Ширванская |
Западная |
|
|
b+-IVS-1-110(G-А) |
30,77 |
6,15 |
13,85 |
|
β0-codon8(-AA) |
16,92 |
3,08 |
- |
|
β0- IVS-2-1(G-A) |
13,85 |
- |
6,15 |
|
b+-IVS-1-5(G-S) |
6,15 |
- |
- |
|
β+-IVS-1-6(T-S) |
3,08 |
- |
- |
Среди талассемиков у 22-х установлены гетерозиготный, 11-ти - гомозиготный, 13-ти компаундный генотипы (рис. 2).
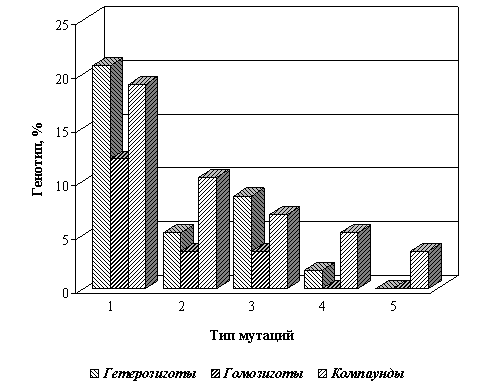
Рис. 2. Участие β-ГГ в формировании генотипов по β-талассемии
1. IVS-1-110(G-A); 2. Codon 8(-AA); 3. IVS-2-1(G-A); 4. IVS-1-5(G-S); 5. IVS-1-6(T-S)
Анализ определения активности фермента Г6ФД, проведенный в Муганской зоне среди 40 новорожденных с диагнозом ГБН, позволил установить гемизиготный генотип у 34 больных мальчиков, 22 из которых имели полный дефицит фермента с фенотипом Г6ФД «0», остальные - частичную недостаточность с фенотипом Г6ФД «+». У больных девочек установлен гетерозиготный генотип по исследуемому заболеванию. Идентифицировано 34 больных с диагнозом β-талассемия, среди которых у 17 установлен гетерозиготный генотип по b+-IVS-1-110(G-А), β0-codon8(-AA), β0-IVS-2-1(G-A) и b+-IVS-1-5(G-S) мутациям, у 8-ми -гомозиготный - по b+-IVS-1-110(G-А), β0-codon8(-AA) и β0-IVS-2-1(G-A) мутациям, у 9-ти - компаундность с участием всех пяти типов мутаций b-ГГ. Согласно спискам ВТЭК ЦРБ в данной популяции зарегистрировано 14 мальчиков с диагнозом гемофилия. В Ширванской зоне у 16 новорожденных с диагнозом ГБН, как у мальчиков, так и у девочек установлен полный и частичный дефицит фермента Г6ФД. В ходе исследований выявлено двое компаундов с фенотипами β+-IVS-1-110(G-А)/β0-codon 8(-АА) и один гомозиготный больной - b+-IVS-1-110(G-А)/b+-IVS-1-110(G-А). Установлено 7 мальчиков с гемофилией. В Западной зоне среди 47 детей с ГБН у 25 новорожденных, а именно у 20 мальчиков идентифицирован гемизиготный генотип и нулевая и частичная активность энзима. Обследованные девочки - гетерозиготы по данному заболеванию. Идентифицировано 9 больных с β-талассемией, среди которых пятеро являлись гетерозиготами по β+-IVS-1-110(G-А) и β0-IVS-2-1(G-А) мутациям, двое - гомозиготами - b+-IVS-1-110(G-А)/b+-IVS-1-110(G-А), двое - компаундами - β+-IVS-1-110(G-А)/β0-IVS-2-1(G-А). По спискам ВТЭК ЦРБ среди популяции Западной зоны зарегистрировано 12 мальчиков с гемофилией.
В ходе проведенной работы в трех крупных зонах республики установлено пять типов компаундов со следующими фенотипами: β+-IVS-1-110(G-А)/β0-IVS-2-1(G-А) - четверо больных из Муганской и Западной зон, β+-IVS-1-6(T-S)/β0-codon 8(-АА), β+-IVS-1-5(G-S)/β+-IVS-1-6(T-S) и β+-IVS-1-5(G-S)/β+-IVS-1-110(G-А) - четверо больных из Муганской зоны, β+-IVS-1-110(G-А)/β0-codon 8(-АА) - пять больных из Муганской и Ширванской зон.
Таким образом, проведенные исследования позволили установить высокую частоту распространения β-талассемии, связанной с частичным и полным отсутствием экспрессии β-ГГ, и идентифицировать два фенотипа по дефициту фермента Г6ФД у новорожденных с гемолитической болезнью в популяции Муганской, Ширванской и Западной зон Азербайджана,. Значимость настоящего исследования заключается в изучении молекулярно-генетической характеристики распределения патологических генов недостаточности Г6ФД, гемофилии и β-талассемии среди обследованного населения для разработки программ генетического скрининга беременных и новорожденных с целью выявления брачных пар с риском рождения больных детей, а также новорожденных с различными формами и сочетаниями гемоглобинопатий, что позволяет оказать своевременное лечение и ретроспективно выявлять семьи, имеющие риск повторного рождения подобных детей.
СПИСОК ЛИТЕРАТУРЫ:
- Альпидовский В.К. Курс гематологии кафедры госпитальной терапии: Учебное пособие. М.: Изд-во РУДН, 2002. Режим доступа: http://med.pfu.edu.ru/_new/russian/win/library/gematologia/nasl.htm
- Бойтлер Э. Нарушения метаболизма эритроцитов и гемолитическая анемия: Пер. с англ. - М.: Медицина. 1981. 256 с.
- Бочков Н.П. Клиническая генетика: Учебник. М.: ГЭОТАР-МЕДИА. 2006. 480 с.
- Краснопольская К. Д. Наследственные болезни обмена веществ: справочное пособие для врачей/ М.: Фохат. 2005. 364 с.
- Ли Ч. Введение в популяционную генетику. М.: Мир. 1978. 546 с.
- Плахута Т., Якунина Л. Гемофилия у детей. Газ. «Медицинская газета», 2003, август, №62.
- Талассемия и другие гемоглобинопатии. Доклад Секретариата. Всемирная Организация Здравоохранения, Женева. ЕВ 118/5, пункт 5.2; 4.05.2006 г.
- Шостакович-Корецкая Л.Р., Маврутенков В.В., Братусь Е.В. и др. Полимеразно-цепная реакция: принципы и практические рекомендации по использованию в клинической практике врача (руководство для врачей). Днепропетровск: МЗ Украины. 2002. 26 с.
- Beutler E. G-6-PD: Population genetics and clinical manifestations // Blood Reviews. 1996. 10: 45-52.
- Cao A. A world-wide evaluation of one on going research and future directions toward preventing thalassemia and sickle sell disease / 2nd International Thalassemia Summer School. 01-05 April 2002, Kurenia/North Cyprus. Р. 7-8.
- El-Beshlawy A., Kaddah N., Moustafa A. et al. Screening for β-thalassaemia carriers in Egypt: significance of the osmotic fragility test // Eastern Mediterranean Health Journal. 2007. Vol. 13. No. 4. Р. 780-786.
- Leung T.N., Lau T.K., Chung T.Kh. Thalassaemia screening in pregnancy // Current opinion in obstetrics & gynecology. 2005. Р.129-134.
- Medina M. D., Vaca G., Lopez-Guido B. et al. Molecular Genetics of Glucose-6-Phosphate Dehydrogenase Deficiency in Mexico // Blood Cells, Molecules, and Diseases. 1997. 23(5) Mar 15. Р. 88-94.
- Modell B. The ethics of prenatal diagnosis and genetic counseling // World Health Forum. 1990. 11. Р. 179.
- Morengo-Rowe A.J. Rapid electroforesis on cellulose acetate. // J. Clin. Pathology. 1965. 18. P. 790.
- Rasulov E. Express-methods of hemoglobinopathy dignosis in newborns // Journal of molecular genetics, microbiology, virusology. 1990. 1. P. 27.
- Saiki R. K., Scharf S., Faloona F. et al. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. // Science. 1985. 230. Р. 1350.
Библиографическая ссылка
Акперова Г.А АНАЛИЗ НАСЛЕДСТВЕННЫХ НАРУШЕНИЙ СИСТЕМЫ КРОВИ В ТРЕХ ЗОНАХ АЗЕРБАЙДЖАНА // Фундаментальные исследования. 2008. № 7. С. 14-18;URL: https://fundamental-research.ru/en/article/view?id=3434 (дата обращения: 16.07.2026).