


Scientific journal
Fundamental research
ISSN 1812-7339
"Перечень" ВАК
ИФ РИНЦ = 1,798
SYNTHETIC APPROACHES TO THE OBTAINING OF ASYMMETRICAL BIS-SPIROPYRANS WITH NAPHTOXAZINONE SITE

Фотохромные соединения способны претерпевать существенные изменения спектров поглощения под действием активирующего облучения. Сопутствующие изменения структуры, дипольного момента, электронного распределения и прочих характеристик позволяют создавать на основе принципа фотохромной реакции функциональные материалы для оптических элементов, управления молекулярной электроникой, хранения оптической информации, компьютерно-генерируемой голографии и т.д. [4, 6]. Наличие в структуре молекулы двух фотохромных центров увеличивает количество потенциально достижимых фотоинициируемых изомеров до четырех (схема) и расширяет перспективы применения таких бис-фотохромных соединений в качестве молекулярных переключаемых свойств.
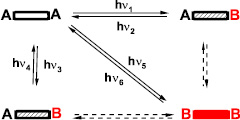
Общая схема изомеризации бис-спиропиранов
В настоящей работе исследован подход к получению систематических серий несимметричных бис-спиропиранов соединений с различными гетареновыми узлами, объединенными общим пиранохроменовым фрагментом.
Цель работы – разработка стратегии получения на основе орто-дигидрокси-диформил-ароматических соединений серий несимметричных бис-спиропиранов, отличающихся как расположением, так и природой гетареновых фрагментов.
Результаты исследования и их обсуждение
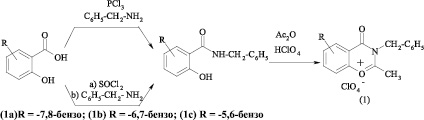
Исходные соли оксазинония (1) получены на основе амидов соответствующих оксинафтойных кислот, которые в свою очередь синтезированы по двум методикам:
– посредством взаимодействия с SOCl2 и последующей обработкой хлорангидрида кислоты соответствующими аминами;
– посредством взаимодействия с реакционно-активным комплексом на основе PCl3 и соответствующих аминов.
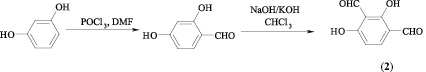
Исходный 2,4-дигидрокси-изо-фталевый альдегид (2) получен последовательным формилированием резорцина по методу Вильсмайера и Реймера – Тимана.
Нафтоксазиноновыйспиропирансбензильным заместителем у атома азота (3а) был получен путем реакции соответствующей соли нафтоксазин-1,3-ония (1a) с 2,4-дигидрокси-изо-фталевым альдегидом (2) и последующей обработкой полученного стирильного производного органическим основанием. Используя (3а) в качестве аналога салицилового альдегида в реакции с перхлоратом 1,2,3,3-триметилиндоленилия в присутствии пиперидина, получен несимметричный индолино-нафтоксазино-бис-спиропиран (4a).
Вторая опробованная стратегия синтеза бис-спиропирана (4а) состояла в конструировании индолиновогогетаренового фрагмента при реакции альдегида (2) с перхлоратом 1,2,3,3-тетраметилиндоленилия в присутствии пиперидина и последующей реакции полученного индолино-спиропирана (5) с солью оксазинония (1а) в уксусной кислоте. Однако последняя стадия не приводила к получению достаточных количествах стирильной соли (6), а попытка проведения реакции в спирте в присутствии пиперидина привела к получению большого количества побочных продуктов, что в итоге не позволило произвести синтез бис-спироструктуры (4а) по альтернативной стратегии.
При апробации указанных стратегий проявлялась ранее исследованная селективность в реакциях альдегида (2) с различными органическими солями, что объясняется совокупностью стерических факторов и приводит к различному расположению гидроксильной и альдегидной групп в спиропиранах (3а) и (5) [1, 2, 3, 5].
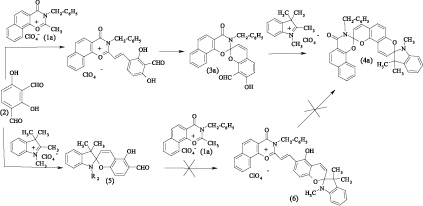
Спиропираны (3b,c) и бис-спиропираны (4b,c) были получены и выделены в виде смеси изомеров (3b-a, 3b-b), (3c-a, 3c-b) и (4b-a, 4b-b), (4c-a, 4c-b) из-за отсутствия упомянутой селективности конденсации по неэквивалентным и конкурирующим альдегидным группам в молекуле исходного альдегида (3).
Структура полученных соединений и состав смесей изомеров (3b-a, 3b-b), (3c-a, 3c-b) и (4b-a, 4b-b), (4c-a, 4c-b) была подтверждена с использованием элементного анализа, ИК-и ЯМР 1Н спектроскопии.
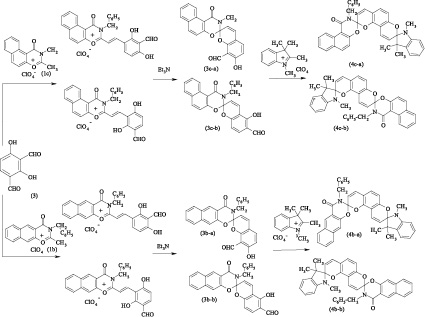
Разделение изомеров методом колоночной хроматографии позволит получить несимметричные бис-спиропираны (4b-a), (4b-b), (4c-a) и (4c-b), характеризующиеся не только неэквивалентной структурой гетареновых структурных узлов, но и различным относительным расположением фотоактивных центров в молекуле.
Заключение
Были разработаны подходы к получению несимметричных бис-спиропиранов, отличающихся не только строением, но и взаимным расположением спиропирановых фрагментов в одной молекуле. Дальнейшее исследование фотохромных свойств полученных соединений позволит проанализировать взаимное электронное влияние взаимной ориентации фотоактивируемых узлов в молекулах бис-спиропиранов.
Экспериментальная часть
ИК-спектры поглощения регистрировались на инфракрасном Фурье-спектрометре (Excalibur HE 3100, PC). Съемка ИК-спектров осуществлялась с помощью метода нарушенного внутреннего полного отражения. Съемка спектров ЯМР 1Н проводилась на радиоспектрометре Bruker 250 (250 MГц) в импульсном Фурье-режиме в дейтерохлороформе. Положение сигналов исследуемого вещества определялось по δ–шкале, отнесение сигналов проведено относительно остаточных сигналов протона дейтерорастворителя – дейтерохлороформа, константа спин-спинового взаимодействия J дана в герцах.
Общая методика получения 3-бензил-2,3-дигидронафто[1,3]-оксазин-4-оксо-2-спиро-2′H,8′H-пирано-[2,3-f]хромен-8′-спиро-2′′-1′′,3′′,3′′-триметилиндолинов (4) N-Бензиламид 1-гидрокси-2-нафтойной кислоты
а) В трехгорлую круглодонную колбу, снабженную механической мешалкой и обратным холодильником, поместили 10 г бензиламина и 40 мл пиридина, при охлаждении и перемешивании по каплям добавили раствор 1,3 мл треххлористого фосфора в 7 мл пиридина. Перемешивали в течение 30 мин без охлаждения, после чего добавили 5,3 г 1-гидрокси-2-нафтойной кислоты и нагревали на водяной бане в течение 4 часов. Отфильтровывали выпавший осадок полифосфорной кислоты. Раствор упарили. Остаток обработали смесью EtOAc и HCl (2:1), так чтобы рН водного слоя составлял 3–4. Собирали органическую фазу, промыли 10 %-м р-ром NaHCO3, а затем водой; отгоняли этилацетат. Выпавший осадок N-бензиламида 1-гидрокси-2-нафтойной кислоты перекристаллизовали из этилацетата/гексана (1/1). Выход 4 г (51 %).
б) В круглодонную колбу на 0,5 л, снабженную капельной воронкой и обратным холодильником, поместили 56,4 г 1-гидрокси-2-нафтойной кислоты, 150 мг карбамида и 200 мл толуола. Прибавили по каплям эквимолярное количество тионилхлорида и кипятили до полного прекращения выделения HCl. Затем к полученному раствору добавили 0,6 М бензиламина. Отфильтровали от осадка бензиламина солянокислого. Толуол отгоняли, выпавшие темно-желтые кристаллы N-бензиламида 1-гидрокси-2-нафтойной кислоты отфильтровывали и перекристаллизовывали из этилацетата/гексана (1/1). Выход 15,2 г (~25 %).
Перхлорат 2-метил-3-бензил-2,3-дигидронафто[2,1-e][1,3]оксазин-4-ония (1а)
К раствору 2,77 г (0,01 М) N-бензиламида 1-гидрокси-2-нафтойной кислоты в 6 мл уксусного ангидрида по каплям при охлаждении добавили 1 мл хлорной кислоты (72 %). Реакционную смесь осторожно, не доводя до кипения, нагревали в течение 5 минут. Выпавший перхлорат 3-бензил-2-метил-2,3-дигидронафто[2,1-e][1,3]оксазин-4-ония (19) фильтруют, промывают эфиром. Выход 2,4 г (60 %).
3-бензил-7′-гидроксил-8′-формил-2,3-дигидронафто[2,1-e][1,3]-оксазин-4оксо-2-спиро-2′-бензопиран (3а)
Кипятили смесь 1,66 г (0,01 М) 2,4-дигидрокси-изо-фталевого альдегида (2) и 4,01 г (0,01 М) перхлората 3-бензил-2-метил-2,3-дигидронафто[2,1-e][1,3]оксазин-4-ония (1а) в 10 мл уксусной кислоты. Охладили реакционную смесь, выпавший осадок отфильтровали и промыли абсолютным эфиром. Поместили в колбу с 30 мл абсолютного эфира и прилили 1,5 мл триэтиламина. Через двое суток эфир декантировали, упарили, и выпавший осадок спиропирана (3a) перекристаллизовали из этилового спирта. Выход 1,44 г (32 %).
3-Бензил-2,3-дигидронафто[2,1-e][1,3]-оксазин-4-оксо-2-спиро-2′H,8′H-пирано-[2,3-f]хромен-8′-спиро-2′′-1′′,3′′,3′′-триметилиндолин (4a)
В колбу поместили 0,449 г (0.001 М) 3-бензил-7′-гидроксил-8′-формил-2,3-дигидронафто[2,1-e][1,3]-оксазин-4-оксо-2-спиро-2′-бензопиран (3а) и 0,274 г (0,001 М) перхлората 1,2,3,3-тетраметилиндоленилия в 10 мл изопропилового спирта. Прилили по каплям при нагревании 0,1 мл (0,0011 М) пиперидина. Далее кипятили реакционную смесь в течении десяти минут. Осадок бисспиропирана (4a) отфильтровали и перекристаллизовали из гексана. Выход 0,12 г (20 %).
3-Бензил-2,3-дигидронафто[1,3]-оксазин-4-оксо-2-спиро-2′H,8′H-пирано-[2,3-f]хромен-8′-спиро-2′′-1′′,3′′,3′′-триметилиндолины (4b,c), выделенные в виде смесей изомеров получены по аналогичной методике на основе 2,3- и 2,1-гидроксинафтойной кислоты.
Работа получила финансовую поддержку Федеральной целевой программы «Научные и научно-педагогические кадры инновационной России» на 2009–2013 годы (соглашение14.А18.21.1188 и № 14.A18.21.0796).
Работа выполнена при финансовой поддержке грантов РФФИ (грант № 12-03-31455 мол_а).
Рецензенты:
Черныш Ю.Е., д.х.н., старший научный сотрудник, Научно-исследовательский институт физической и органической химии Южного федерального университета, г. Ростов-на-Дону;
Стариков А.Г., д.х.н., ведущий научный сотрудник, Научно-исследовательский институт физической и органической химии Южного федерального университета, г. Ростов-на-Дону.
Работа поступила в редакцию 17.10.2013.
Библиографическая ссылка
Ожогин И.В., Муханов Е.Л., Комиссарова О.А., Лукьянова М.Б., Дороган И.В., Лукьянов Б.С. СИНТЕТИЧЕСКИЕ ПОДХОДЫ К ПОЛУЧЕНИЮ НЕСИММЕТРИЧНЫХ БИС-СПИРОПИРАНОВ С НАФТОКСАЗИНОНОВЫМ ФРАГМЕНТОМ // Фундаментальные исследования. 2013. № 10-9. С. 1973-1977;URL: https://fundamental-research.ru/en/article/view?id=32570 (дата обращения: 24.06.2026).