


Scientific journal
Fundamental research
ISSN 1812-7339
"Перечень" ВАК
ИФ РИНЦ = 1,798
TOTAL SYNTHESIS OF NATURAL PHENOLGLYCOSIDE SALICYLOYL-SALICINE AND ITS ANALOG SALICYLOY-SALIREPIN

Фенолгликозиды широко распространены в растениях семейства Salicaceae (Ивовые). Фитопрепараты, изготовленные из различных частей растений семейства Ивовые [3], хорошо зарекомендовали себя при лечении целой гаммы заболеваний, таких как болезни легких, натуральная оспа, артрит, в народной медицине они используются как эффективные жаропонижающие и противовоспалительные средства [5]. Одним из наиболее распространенных соединений в растениях семейства Ивовые наряду с салицином и салирепозидом (2-(β-D-глюкопиранозилокси)-5-гидрокси-бензилбензоат) является салициоил-салицин [8, 1]. Это соединение интересно с медицинской точки зрения, так как содержит остаток салициловой кислоты, которая, в свою очередь, обладает широким спектром биологической активности [6,7].
Таким образом, цель данного исследования заключается в разработке методик синтеза природного фенолгликозида салицилоил-салицина и его гидроксилированного аналога – салицилоил-салирепина, а также выявление данных соединений в составе экстрактивных веществ коры осины.
Результаты исследования и их обсуждение
Синтез осуществлялся следующим образом (схема). Гликозид 1 был получен согласно методике, описанной ранее [6], при гликозилировании ацетил-гентизинового альдегида с последующим восстанавлением альдегидной группы до спиртовой. Гликозид 2 был получен при гликозилировании орто-крезола с последующим бромированием [2]. Свободная спиртовая группа гликозида 1 ацилировалась хлорангидридом ацетилсалициловой кислоты с получением гликозида 3. В случае гликозида 2 использовалась ацетилсалициловая кислота с получением соединения 4 или салициловая кислота с получением гликозида 5. Примечательно, что последняя реакция была ранее осуществлена еще в 1959 в лаборатории Земплена [4]. Однако мы обнаружили несовпадение температуры плавления нашего соединения (93–94 °С) и соединения, полученного Земпленым (163 °С). Структура нашего образца 5 была подтверждена методом ЯМР-спектроскопии, а чистота также подтверждена методами ВЭЖХ и ГХ-МС, в то время как в работе [4] приводится только элементный анализ поэтому, целесообразно полагать, что физико-химические характеристики нашего образца представлены более точно.
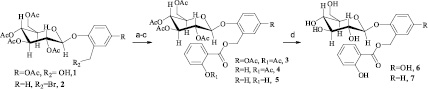
Синтез салицилоил-салицина и салицилоил-салирепинаа.a Реагенты и условия: (a) ацетил салицилоилхлорид, пиридин, CHCl3, 24ч, RT; (b) ацетилсалициловая кислота, NaHCO3, ДМФА, 24ч, RT; (c) салициловая кислота, NaHCO3,ДМФА, 24 ч, RT; (d) 36 % HCl, CHCl3, EtOH, 30 °C, 8 ч
Кроме того, недостатком методики Земплена является то, что ему так и не удалось получить целевой гликозид салицилоил-салицин 7, так как при снятии защитных групп отщеплялся также остаток салициловой кислоты. Нам же успешно удалось провести селективное снятие ацетильных групп без существенного гидролиза сложноэфирной салицилоильной связи, и гликозид 7 был получен с довольно высоким выходом (75 %). Также путем снятия ацетильных групп в присутствии салицилоильной из полного ацетата 3 был получен салицилоил-салирепин 6 (выход 65 %).
Исследование состава коры осины методами хроматомассспектрометрии, высокоэффективной жидкостной хроматографии и ТСХ в сравнении со стандартными образцами фенолгликозидов 6, 7 показало наличие салицилоил-салицина и салицилоил-салирепина (рис. 1–3).
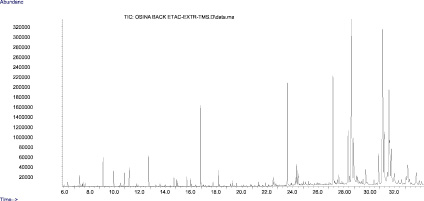
Рис. 1. Хроматограмма экстракта коры осины (в виде ТМС-производных) по общему ионному току
Адекватного хроматографического разделения салирепозида (одного из основных фенолгликозидов коры осины) и салицилоил-салицина 7 удалось достичь только при использовании ацетилированных производных (рис. 3). При разделении этой пары фенолгликозидов в виде ТМС-производных наблюдалось очень близкое время удерживания – 27.757 мин (салицилоил-салицин 7) и 27.782 мин (салирепозид), что не позволило достоверно судить о наличии гликозида 7.
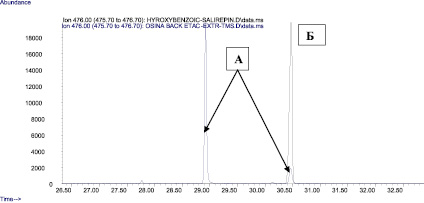
Рис. 2. Совмещенные хроматограммы экстракта коры осины (в виде ТМС-производных) по 476 иону (А) и ТМС-производное салицилоил-салирепина по 476 иону (Б)
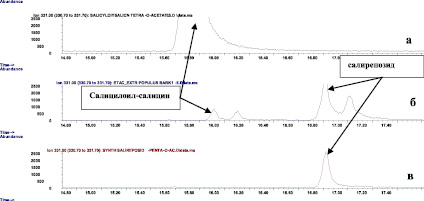
Рис. 3. а – хроматограмма стандарта – пента-О-ацетил-салицилоил-салицина по 331 иону; б – хроматограмма ацетилированного экстракта осины по 331 иону; в – хроматограмма пента-О-ацетилсалирепозида по 331 иону
Таким образом, нами впервые был получен природный фенолгликозид салицилоил-салицин 7, а также его аналог, салицилоил-салирепин 6, обладающий потенциальной биологической активностью. Количественное определение гликозида 6 в экстракте коры осины было проведено методом ВЭЖХ с использованием в качестве внутреннего стандарта резорцина. Содержание гликозида 6 составило 9 ± 3 % в экстракте коры осины или в среднем 0,41 % в пересчете на воздушно-сухую кору.
Фенолгликозид 6 впервые обнаружен в коре осины. До настоящего момента это первый факт существования соединения такой структуры в растительных организмах.
Материалы и методы исследования
1. Приборы и оборудование
Контроль за ходом реакции и чистотой полученных продуктов вели методом ТСХ на пластинках Silufol-UV 254 и Kieselgel 60 F254. Детектирование пятен проводили в фильтрованном УФ-свете. В качестве элюента использовалась система бензол:этанол (10:1), хлороформ:этанол (4:1). ВЭЖХ – анализ проводился на жидкостном хроматографе Agilent Compact LC с колонкой 150×4.6 с неподвижной фазой Exlips Plus C-18 (5 мкм). Анализ проводили посредством градиентного элюирования смесью ацетонитрил–вода с добавлением 0,1 % трифторуксусной кислоты как модификатора подвижной фазы. Условия градиента: 0 % ацетонитрила при 0, 40 мин – до 20 % ацетонитрила, 70 мин – до 40 % ацетонитрила при скорости потока 1 мл/мин (А), и 0 % ацетонитрила при 0, 20 мин – до 100 % (Б). Проба в объеме 20 мкл. Детектирование осуществляли при 220 и 276 нм (салицилоил-салицин).
Газовую хроматографию с масс-детектированием проводили с использованием квадрупольного масс – детектора Agilent 5975 C и газового хроматографа Agilent 7890A. Энергия ионизации 70 эВ, температура ионного источника 230 °С, квадруполя 150 °С, испарителя – 280 °С, интерфейса 290 °С. Объем вводимой пробы 1 мкл при делении потока 1:5. Капиллярная колонка НР-1 MS 30 м×0,25 мм×0,25 мкм (Agilent). Диапазон сканирования масс – 33–600 а.е.м. Программирование температуры: 2 мин при 70 °C, 70–315 °С (10 °С/мин), 15 мин при 315 °С. Газ носитель ‒ гелий, скорость потока 1 мл/мин. Дериватизацию осуществляли силилированием гексаметилдисилазаном в пиридине в присутствии трифторуксусной кислоты (получение ТМС-производных) и ацетилированием в пиридине (получение полностью ацетилированных производных) по известным методикам [10].
Температуру плавления веществ определяли в капилляре с использованием MP50 Melting point system (Mettler toledo). УФ-спектры снимали на спектрофотометре СФ-102. ИК-спектры снимали на ИК-Фурье спектрометре Spectrum BX II в таблетках бромистого калия. 1Н и 13С – ЯМР спектры снимали на приборе Bruker-300 MMX (300 и 80 МГц).
2. Растительный материал: кора Populus tremula была собрана в окрестностях г. Томска в мае 2011 г.
3. Выделение суммарной фракции фенолгликозидов. Воздушно-сухую кору осины (50 г) измельчали до кусков размером 5–8 мм и экстрагировали кипящим 70 % водным этанолом (3×200 мл) в течение 30 минут. После удаления основного количества этанола под уменьшенным давлением при 60 °С остаток был суспендирован в 100 мл воды, экстрагирован однократно гексаном (50 мл) и этилацетатом (6×50 мл). После удаления этилацетата было получено 2,3 г остатка (4,6 %).
4. 2-(2,3,4,6-тетра-О-ацетил-β-D-глюкопиранозилокси)-5-ацетилоксибензил (2-ацетокси)бензоат (гексаацетат салицилоил-салирепина) (3). К 0,2 моль гликозида 1, растворенного в 1 мл сухого хлороформа, добавляли 0,22 ммоль ацетил-салицилоилхлорида и 0,26 ммоль пиридина. Реакционную массу выдерживали при н. у. в течение 24 ч, после чего добавляли 20 мл CHCl3, промывали раствор 0,1 М H2SO4, насыщенным раствором Na2CO3, водой, сушили над Na2SO4 и отгоняли CHCl3 под вакуумом. Остаток кристаллизовали из этилового спирта. Выход 50 %. Т.пл. 103–104 °С. УФ λmax (EtOH)/нм: 275. ИК (KBr, νmax/cm–1): 1748; 1641; 1604; 1498; 1369; 1214; 1190; 1034; 906; 1Н ЯМР (CDCl3, 300 MHz) δ: 2,02; 2,04; 2,07; 2,010; 2,17; 2,28 с (6×3H, COCH3); 3,81–3,85 м (1Н, Н-5’); 4,14 дд (1Н, J = 2,1; 12,3 Гц, Н-6’b); 4,24 дд (1Н, J = 5,4; 12,3 Гц, H-6’а); 5,00 м (1Н, Н-1’); 5,13–5,18 м (1Н, H-4); 5,23–5,36 м (4H, H-2’, H-3’, 2xН-7); 6,97 дд (1Н, J = 2,7; 8,1 Гц, Н-3); 7,08–7,13 м (3Н, Н-2, H-5, H-13); 7,30 т (1Н, J = 7,5 Гц, Н-11); 7,55 т (1Н, J = 6,9 Гц Н-12); 8,04 д (1Н, J = 7,8 Гц, Н-14), 13C ЯМР (CDCl3, 75,5 MHz) δ: 20,6; 20,09 (6×CН3, COCH3); 61,2 (СН2, С6’); 61,9 (CH2, С-7); 68,3 (СН, С-4’); 71,0 (СН, С-2’); 72,1 (СН, С-3’); 72,6 (СН, С-5’); 99,8 (СН, С-1’); 117,5 (СH, С-2); 122,4 (2xСН, С-3, C-5); 122,8 (C, C-9); 123,9 (CH,C-13); 126,1 (СН, С-11); 128,0 (С, С-6); 131,9 (CH, C-14); 134,0 (CH, C-12); 146,1 (С, С-4); 150,2 (С, С-10); 151,4 (C, C-1); 164,0 (C = O, C-8), 169,3; 169,4; 169,6; 169,7; 170,2; 170,5 (6×С = O, COCH3).
5. Ацилирование гликозида 2. Общая методика. К 0,150 г (0,29 ммоль) гликозида 2, добавляли 0,35 ммоль соответствующей кислоты, 0,35 ммоль гидрокарбоната натрия и 1 мл ДМФА. Перемешивали при 20 °С 24 ч., после чего реакционную массу выливали в 5 мл воды и перемешивали до образования осадка. Осадок отфильтровывали и перекристаллизовывали из этилового спирта.
5.1 2-(2,3,4,6-тетра-O-ацетил-β-D-глюкопиранозилокси)-бензил 2-ацетоксибензоат (пентаацетат салицилоил-салицина) (4) был получен из ацетилсалициловой кислоты. Выход 65 %, Т.пл. 81–82 °C. УФ λmax (EtOH)/нм: 273. ИК (KBr, νmax/cm–1): 30445; 1755; 1608; 1376; 1235; 1072; 1045; 909; 756. 1H ЯМР (CDCl3, 300 MГц) δ: 2,03; 2,04; 2,06; 2,09; 2,11 с (5×3H, COCH3); 3,83 м (1Н, Н-5’); 4,15 дд (1Н, J = 1,9, 12,3 Гц, Н-6’b); 4,25 дд (1Н, J = 5,7; 12,3 Гц, H-6’а); 5,01–5,20 м (3Н, Н-1’, H-4’, H-7b); 5,25–5,30 м (3Н, H-2’, H-3’, Н-7a); 7,00 д (1Н, J = 9,0 Гц, Н-11); 7,03 м (2Н, Н-2, H-4); 7,28 м (2Н, Н-3, Н-13); 7,35 д (1Н, J = 7,8 Гц Н-5); 7,52 тд (1Н, J = 1,5, 7,6 Гц, Н-12); 8,05 дд (1Н, J = 7,8, 1,5 Гц, Н-14). 13C NMR (CDCl3, 75,5 MГц) δ: 20,6 (5×CН3, COCH3); 61,6 (СН2, С6’); 61,8 (CH2, С-7); 68,2 (СН, С-4’); 70,9 (СН, С-2’); 71,9 (СН, С-3’); 72,6 (СН, С-5’); 99,3 (СН, С-1’); 116,1 (СH, С-2); 123,6 (СН, С-4); 123,7 (C, C-9); 123,8 (CH,C-13); 126,0 (СН, С-11); 129,5 (С, С-3); 129,7 (СH, С-5); 130,8 (С, С-6); 131,9 (CH, C-14); 133,9 (CH, C-12); 150,8 (С, С-10); 154,9 (C, C-1); 164,2 (C = O, C-8), 169,3; 170,1; 170,5 (4×С = O, COCH3).
5.2 2-(2,3,4,6-тетра-O-ацетил-β-D-глю-копиранозилокси)-бензил 2-гидроксибензоат (тетраацетатл салицилоил-салицина) (5) был получен из салициловой кислоты. Выход 60 %, Т.пл. 93–94 °C лит. 163 [8]. УФ λmax (EtOH)/нм: 235; 276; 302. ИК (KBr, νmax/cm–1): 1754; 1681; 1487; 1236; 1083; 1047; 908; 762. 1H ЯМР (CDCl3, 300 MГц) δ: 2,03; 2,05; 2,06; 2,07 с (4×3H, COCH3); 3,85 м (1Н, м, Н-5’); 4,14 дд (1Н, J = 2,1, 12,0 Гц, Н-6’b); 4,25 дд (1Н, J = 5,1, 12,3 Гц, H-6’а); 5,11 м (2Н, Н-4’, H-2’); 5,28 м (3Н, СН2,Н-7 H-3’); 5,41 д (1Н, J = 12,9 Гц, H-1’); 6,86 м (1Н, Н-13); 6,96 д (1Н, J = 8,1 Гц, Н-11); 7,08 м (2Н, Н-2, Н-4); 7,30 м (1Н, Н-3); 7,42 м (2Н, Н-5, Н-12); 7,87 д (1Н, J = 6,9 Гц, Н-14). 13C NMR (CDCl3, 75,5 MГц) δ: 20,5 (4×CН3, COCH3); 61,7 (2хСН2, С6’, С-7); 68,2 (СН, С-4’); 70,9 (СН, С-2’); 72,0 (СН, С-3’); 72,5 (СН, С-5’); 99,1 (СН, С-1’); 112,3 (С, С-9); 115,6 (СН, С-2); 117,5 (CH,C-13); 119,1 (CH, C-11); 123,5 (СН, С-4); 125,5 (С, С-6); 129,4 (СH, С-5); 129,7 (СН, С-3); 129,9 (CH, C-14); 135,8 (CH, C-12); 154,4 (С, С-1); 161,6 (C, C-10); 169,2 (C = O, C-8), 169,3; 169,7; 170,2; 170,5 (4×С = O, COCH3).
6. Селективное снятие ацетильных групп. Общая методика. К 0,15 ммоль гликозида, растворенного в смеси этанол-хлороформ в пропорции 1,5–0,5 мл было добавлено 0,5 мл 36 % HCl. Реакционная масса выдерживалась при 30 °С 8 ч (контроль ВЭЖХ), смесь растворителей выпаривалась под вакуумом (температура бани не более 50 °С). Остаток очищали с помощью колоночной хроматографии при градиентном элюировании CHCl3-EtOH (от 15:1 до 8:1). Кристаллизовали из спирта или ацетона.
6.1. 2-(β-D-глюкопиранозилокси)-5-гидрокси-бензил 2-гидроксибензоат (салицилоил-салирепин) (6). Выход 65 %, Т.пл 164–168 °C. UV λmax (EtOH)/nm: 298. IR (KBr, νmax/cm–1): 3480; 3350; 2920; 1670; 1610; 1490; 1210; 1080; 1050; 755. 1H ЯМР (DMSO-d-6, 300 MГц) δ, м.д.: 3,22 м (1Н, Н-4’); 3,68 м (1Н, H-6’а); 4,63 м (1Н, Н-1’); 5,41 м (2Н, H-7); 6,68 дд (1Н, J = 2,7; 8,4 Гц, Н-3); 6,76–6,81 м (2Н, Н-2, H-5,); 6,94 м (2Н, Н-11, H-13); 7,51 т (1Н, J = 7,5 Гц Н-12); 7,83 д (1Н, J = 7,8 Гц, Н-14). В спектре отсутствуют сигналы H-2’, H-3’, H-5’, H-6’b, т.к. перекрываются сигналом растворителя13C NMR (DMSO-d-6, 75,5 MHz) δ: 60,9 (СН2, С6’); 61,9 (CH2, С-7); 69,9 (СН, С-4’); 73,4 (СН, С-2’); 76,5 (СН, С-3’); 77,0 (СН, С-5’); 102,9 (СН, С-1’); 113,0 (СH, С-9); 114,8 (C-5); 115,4 (СН, С-3); 117,3 (CН, C-13); 117,9 (CH, C-2); 119,5 (СН, С-11); 126,0 (С, С-6); 130,1 (CH, C-14); 135,8 (CH, C-12); 148,0 (С, С-1); 152,2 (С, С-4); 160,0 (C, C-10); 168,5 (C = O, C-8).
6.2. 2-(β-D-глюкопиранозилокси) – бензил 2-гидроксибензоат (салицилоил-салицин) (7). Выход 75 %, Т.пл 164–165 °C. UV λmax (EtOH)/nm: 276, 299 (277, 305 лит [3]). IR (KBr, νmax/cm–1): 3391; 2926; 1672; 1615; 1301; 1250; 1084; 756. 1H ЯМР (DMSO-d-6, 300 MГц) δ: 3,16 м (1Н, Н-4’); 3,23 м (2Н, Н-2’, 4’); 3,46 м (2Н, H-6’а, H-5’); 3,68 м (1Н, Н-6’b); 4,86 м (1Н, Н-1’); 5,46 м (2Н, H-7); 6,91 м (2Н, Н-11, H-13); 7,03 т (1Н, J = 7,4 Гц, H-4); 7,18 д (1Н, J = 8,1, Гц, Н-2); 7,31 т (1Н, J = 7,2; 8,1 Гц Н-3); 7,42 д (1Н, J = 7,2 Гц, Н-5); 7,50 м (1H, H- 12); 7,81 д (1Н, J = 8,1, Гц, Н-14). 13C NMR (DMSO-d-6, 75,5 MHz) δ: 60,7 (CH2, C-6’); 62,1 (CH, C-7); 69,6 (CH, C-4’); 73,2 (CH, C-2’); 76,4 (CH, C-3’); 77,1 (CH, C-5’); 101,1 (CH, C-1’); 113,1 (C, C-9); 115,2 (CH, C-2); 117,3 (CН, C-11); 119,5 (CH, C-13); 121,9 (СН, С-4); 124,5 (С, С-6); 129,0 (CH, C-5); 129,7 (СН, C-3); 130,2 (CH, C-14); 135,7 (CН, C-12); 155,3 (C, C-1); 160,0 (C, C-10); 168,6 (C, COO, C-8).
Работа выполнена в рамках государственного задания «Наука» по теме 3.2702.2011.
Рецензенты:
Иванчина Э.Д., д.т.н., профессор кафедры ХТТиХК, ФГБОУ ВПО «Национальный исследовательский Томский политехнический университет», г. Томск;
Ивашкина Е.Н., д.т.н., доцент кафедры ХТТиХК, ФГБОУ ВПО «Национальный исследовательский Томский политехнический университет», г. Томск.
Работа поступила в редакцию 01.07.2013.Библиографическая ссылка
Степанова Е.В., Белянин М.Л. ПОЛНЫЙ СИНТЕЗ ПРИРОДНОГО ФЕНОЛГЛИКОЗИДА САЛИЦИЛОИЛ-САЛИЦИНА И ЕГО АНАЛОГА САЛИЦИЛОИЛ-САЛИРЕПИНА // Фундаментальные исследования. 2013. № 8-3. С. 736-740;URL: https://fundamental-research.ru/en/article/view?id=31992 (дата обращения: 30.06.2026).