


Scientific journal
Fundamental research
ISSN 1812-7339
"Перечень" ВАК
ИФ РИНЦ = 1,798
NEW SYNTHETIC ROUTES FOR DESIGN OF CHEMOSENSOR MATERIALS

Ионактивные системы (хромогенные, флуорогенные и фотохромные) получили широкое распространение как точный инструмент для исследования состава абиотических и биотических объектов. Основными областями применения сенсорных материалов в настоящее время являются контроль качества пищевых продуктов, диагностика некоторых заболеваний, определение чистоты лекарственных препаратов и мониторинг окружающей среды. Наиболее распространенными и чувствительными методами исследования свойств сенсорных систем являются УФ-спектроскопия и флуоресцентный анализ. Оптические методы хемосенсорного анализа имеют ряд преимуществ – высокую чувствительность, применимость в широком диапазоне концентраций исследуемых веществ, простоту исполнения и др.
Поскольку важную роль в определении направленности сенсоров и их чувствительности играет тип соединения рецептора и ионофора, одним из важнейших направлений современной хемосенсорики является разработка методов направленной модификации полимерных материалов за счет ковалентного связывания ионактивных молекул с органической подложкой с учетом влияния различных структурных факторов на сенсорные свойства получаемых материалов [4, 5, 7].
Фотодинамические сенсорные системы, включающие в состав рецепторного фрагмента разнообразные азотсодержащие структуры, могут быть использованы не только для обнаружения катионов, но и в качестве высокоэффективных сенсоров на биологически важные анионы моно- и полиосновных кислот [2, 3, 6, 8].
Материалы и методы исследования
Спектры ЯМР 1Н получены на спектрометре Varian Unity 300 (300 МГц) в CDCl3 или DMSO-d6. В качестве внутреннего стандарта использовались остаточные сигналы CHCl3 (δ 7,25 м.д.) и (CH3)2SO (δ 2,50 м.д.). Электронные спектры поглощения сняты на спектрофотометре Varian Cary 100, спектры люминесценции измерены на спектрофлуориметре Varian Cary Eclipse. Температуры плавления определяли в стеклянных капиллярах на приборе ПТП (М). Полноту протекания реакций и индивидуальность полученных соединений контролировали с помощью ТСХ (пластины Silufol U254, элюент – CHCl3, проявление парами йода во влажной камере).
Общая методика синтеза аминов (4а-е). Растворяли 30 ммоль антрацен-9-карбальдегида в 70 мл бутанола, добавляли 0,05 мл ледяной уксусной кислоты и 33 ммоль амина (2б-е) [или 60 ммоль амина (2а)]. Смесь кипятили 4 ч, упаривали на роторном испарителе и охлаждали. Остаток перекристаллизовывали из подходящего растворителя.
К раствору 20 ммоль полученного азометина (3а-е) в этаноле или смеси этанол–ДМФА (4:1) при нагревании (50–60 °С) и перемешивании постепенно добавляли 50 ммоль боргидрида натрия. Смесь перемешивали при данной температуре 1 ч, разбавляли 200 мл горячей воды и избыток боргидрида разлагали добавлением разбавленной уксусной кислоты. Суспензию охлаждали, осадок отфильтровывали и перекристаллизовывали из подходящего растворителя.
Амин (4а) выделяли из реакционной смеси экстракцией хлороформом. Хлороформ отгоняли, полученное густое масло тщательно растирали с 30 мл конц. соляной кислоты. Через 2–3 ч осадок гидрохлорида амина (4а·HCl) отфильтровывали и промывали сухим ацетоном (2×10 мл).
(Антрацен-9-илметил)(2-метилпропил)амин гидрохлорид (4а·HCl). Выход 73 %. Т.пл. 212–213 °С (разл.). Спектр ЯМР 1Н, δ, м.д. (DMSO-d6): 1,32 д (6Н, J 6,6 Гц, 2СН3); 2,24–2,40 м (1Н, СН); 3,97–4,11 м (2Н, СН2); 4,92 д (2H, J 6,0 Гц, CH2), 7,39–7,54 м (4Н, HAr); 8,05 д (2Н, J 8,40 Гц, HAr); 8,44 д (2Н, J 8,4 Гц, HAr); 8,60 с (1Н, HAr); 9,80 уш.с (1H, N + H2).
(S)-(-)-(Антрацен-9-илметил)(1-фенилэтил)амин (4б). Выход 78 %. Т.пл. 76–77 °С. Спектр ЯМР 1Н, δ, м.д. (DMSO-d6): 1,42 д (3Н, J 7,4 Гц, СН3); 4,10 д.д (1Н, J1 4,8 Гц, J2 14,8 Гц, СН); 4,53 д.д (2Н, J1 13,4 Гц, J2 26,8 Гц, СН2); 7,18–7,60 м (9Н, HAr); 7,90–8,20 м (5 Н, HAr, NH); 8,40 c (1Н, HAr).
N-(Антрацен-9-илметил)-2-метоксианилин (4в). Выход 88 %. Т.пл. 183–184 °С. Спектр ЯМР 1Н, δ, м.д. (DMSO-d6): 3,86 c (3Н, СН3); 3,90 ш.с. (1H, NH), 5,10 д (2H, J 5,6 Гц, CH2), 7,10–7,21 м (2Н, HAr); 7,26–7,40 м (2Н, HAr); 7,42–7,60 м (4Н, HAr); 8,02 д (2Н, J 8,8 Гц, HAr); 8,41 д (2Н, J 8,8 Гц, HAr); 8,64 с (1Н, HAr).
N-(Антрацен-9-илметил)-3,5-дихлоранилин (4г). Выход 94 %. Т.пл. 201–202 °С. Спектр ЯМР 1Н, δ, м.д.(DMSO-d6): 4,05 ш.с. (1H, NH), 5,12 д (2H, J 5,4 Гц, CH2), 7,15–7,40 м (3Н, HAr); 7,46–7,62 м (4Н, HAr); 8,04 д (2Н, J 8,4 Гц, HAr); 8,42 д (2Н, J 8,2 Гц, HAr); 8,58 с (1Н, HAr).
2-[(Антрацен-9-илметил)амино]этан-1-ол (4д). Выход 82 %. Т.пл. 118–119 °С. Спектр ЯМР 1Н, δ, м.д.(DMSO-d6): 2,88 т (2Н, J 5,4 Гц, СН2); 3,57 т (2Н, J 4,8 Гц, СН2); 4,29 ш.с (1Н, NH); 4,65 с (2Н, СН2); 7,40–7,56 м (4Н, HAr); 8,01 д (2Н, J 8,1 Гц, HAr); 8,38 д (2Н, J 8,1 Гц, HAr); 8,42 c (1Н, HAr).
3-[(Антрацен-9-илметил)амино]пропан-1-ол (4е). Выход 72 %. Т.пл. 83–84 °С. Спектр ЯМР 1Н, δ, м.д.(DMSO-d6): 1,78 кв (2Н, СH2); 3,14 т (2Н, J 5,7 Гц, СН2); 3,84 т (2Н, J 5,7 Гц, СН2); 4,76 с (2Н, СН2); 7,42–7,61 м (4Н, HAr); 8,02 д (2Н, J 8,4 Гц, HAr); 8,31 д (2Н, J 8,4 Гц, HAr); 8,43 с (1Н, HAr).
Общая методика синтеза амидов (6а-г) и (8б, г). Растворяли 10 ммоль соответствующего амина (4а-г) в 50 мл сухого толуола, добавляли сухой пиридин [20 ммоль для аминов (4б-е) или 40 ммоль для амина (4а)] и при интенсивном перемешивании добавляли раствор 1,25 ммоль свежеперегнанного хлорацетилхлорида (или 3-хлорпропионилхлорида) в 10 мл сухого толуола. Реакционную смесь кипятили в течение 2–3 ч (6в, г и 8г) или перемешивали при комнатной температуре 10 ч (6а, б и 8б). Охлаждали до 5 °С, выпавший осадок гидрохлорида пиридина отфильтровывали, промывали на фильтре толуолом (2×5 мл). Фильтрат промывают водой (2×20 мл) и упаривали на роторном испарителе, остаток после охлаждения перекристаллизовывали из подходящего растворителя.
N-(Антрацен-9-илметил)-N-(2-метилпропил)-2-хлорацетамид (6а). Выход 68 %. Т.пл. 61–62 °С (гексан-бензол – 4:1). Спектр ЯМР 1Н, δ, м.д. (DMSO-d6): 1,40 д (6Н, J 6,2 Гц, 2СН3); 2,20–2,41 м (1Н, СН); 3,70–4,02 м (4Н, 2СН2); 5,24 с (2H, CH2), 7,44–7,64 м (4Н, HAr); 8,03 д (2Н, J 8,6 Гц, HAr); 8,40 д (2Н, J 8,6 Гц, HAr); 8,55 с (1Н, HAr).
(S)-(-)-N-(Антрацен-9-илметил)-N-(1-фенилэтил)-2-хлорацетамид (6б). Выход 75 %. Т.пл. 64–65 °С (гексан-бензол – 3:1). Спектр ЯМР 1Н, δ, м.д. (DMSO-d6): 1,45 д (3Н, J 7,0 Гц, СН3); 3,80 с (2H, CH2); 4,02 д.д (1Н, J1 5,0 Гц, J2 16,0 Гц, СН); 5,21 д (2Н, J 7,2 Гц, СН2); 7,18–7,60 м (9Н, HAr); 7,97–8,20 м (3Н, HAr); 8,33 д (2Н, J 8,8 Гц, HAr).
N-(Антрацен-9-илметил)-N-(2-метоксифенил)-2-хлорацетамид (6в). Выход 81 %. Т.пл. 161–162 °С (толуол). Спектр ЯМР 1Н, δ, м.д. (DMSO-d6): 3,72 с (2H, CH2); 3,94 c (3Н, СН3); 5,92 с (2H, CH2), 7,12–7,25 м (2Н, HAr); 7,30–7,44 м (2Н, HAr); 7,48–7,66 м (4Н, HAr); 8,05 д (2Н, J 8,4 Гц, HAr); 8,37 д (2Н, J 8,4 Гц, HAr); 8,51 с (1Н, HAr).
N-(Антрацен-9-илметил)-N-(3,5-дихлорфенил)-2-хлорацетамид (6г). Выход 84 %. Т.пл. 177–178 °С (тетрагидрофуран). Спектр ЯМР 1Н, δ, м.д. (DMSO-d6): 3,97 с (2H, СH2), 6,05 с (2H, CH2), 7,11–7,41 м (3Н, HAr); 7,46–7,62 м (4Н, HAr); 8,03 д (2Н, J 8,8 Гц, HAr); 8,46 д (2Н, J 8,8 Гц, HAr); 8,60 с (1Н, HAr).
(S)-(-)-N-(Антрацен-9-илметил)-N-(1-фенилэтил)-3-хлорпропионамид (8б). Выход 70 %. Т.пл. 56–57 °С (гексан-бензол – 4:1). Спектр ЯМР 1Н, δ, м.д. (DMSO-d6): 1,42 д (3Н, J 6,8 Гц, СН3); 2,91 д (2H, J 6,8 Гц, CH2); 3,42 д (2H, J 6,8 Гц, CH2); 3,94 д.д (1Н, J1 5,0 Гц, J2 15,0 Гц, СН); 5,04 д (2Н, J 6,8 Гц, СН2); 7,14–7,55 м (9Н, HAr); 7,92–8,17 м (3Н, HAr); 8,40 д (2Н, J 8,6 Гц, HAr).
N-(Антрацен-9-илметил)-N-(3,5-дихлорфенил)-3-хлорпропионамид (8г). Выход 77 %. Т.пл. 172–173 °С (бутанол). Спектр ЯМР 1Н, δ, м.д. (DMSO-d6): 2,96 д (2H, J 7,0 Гц, CH2); 3,55 д (2H, J 7,0 Гц, СH2), 5,88 с (2H, CH2), 7,16–7,40 м (3Н, HAr); 7,50–7,65 м (4Н, HAr); 8,05 д (2Н, J 8,6 Гц, HAr); 8,42 д (2Н, J 8,6 Гц, HAr); 8,52 с (1Н, HAr).
N-(Антрацен-9-илметил)-N-(2-хлорэтил)амин гидрохлорид (9д·HCl). Растворяли при нагревании до 40–45 °С 2,51 г (10 ммоль) спирта (4д) в 50 мл сухого хлороформа и осторожно при перемешивании добавляли 0,9 мл (12 ммоль) свежеперегнанного SOCl2. Полученную смесь кипятили в течение 5–6 ч, упаривали до 1/3 объема и охлаждали. Выпавший осадок гидрохлорида (9д·HCl) отфильтровывали, промывали на фильтре этоксиэтаном (2×5 мл), сушили и кристаллизовали из бутанола. Выход 2,70 г (90 %). Т.пл. 222–223 °С (бутанол). Спектр ЯМР 1Н, δ, м.д. (DMSO-d6): 3,44–3,63 м (2Н, СН2); 4,00 т (2Н, J 6,2 Гц, СН2); 5,26 ш. с (2Н, СН2); 7,50–7,73 м (4Н, HAr); 8,12 д (2Н, J 8,6 Гц, HAr); 8,55 д (2Н, J 8,6 Гц, HAr); 8,71 c (1Н, HAr); 9,85 ш. с (2Н, N + H2).
Основание (9д) выделяли обработкой 10 ммоль гидрохлорида (9д·HCl) 30 мл 10 %-ного раствора Na2CO3. Суспензию тщательно размешивали и экстрагировали основание (9д) хлороформом (3×15 мл), полученный экстракт сушили безводным Na2SO4 и отгоняли растворитель на роторном испарителе при пониженном давлении. Выход 90 %. Полученное вещество являлось хроматографически чистым и могло быть использовано в дальнейших реакциях без дополнительной очистки.
N-(Антрацен-9-илметил)-N-(3-хлорпропил)амин гидрохлорид (9е·HCl). Получали аналогично соединению (4д) хлорированием спирта (4е). Выход 82 %. Т.пл. 200–201 °С (бутанол). Спектр ЯМР 1Н, δ, м.д. (DMSO-d6): 1,90–2,05 м (2Н, СH2); 3,44 т (2Н, J 6,2 Гц, СН2); 4,14–4,25 м (2Н, СН2); 5,18 с (2Н, СН2); 7,38–7,55 м (4Н, HAr); 8,07 д (2Н, J 8,6 Гц, HAr); 8,42 д (2Н, J 8,6 Гц, HAr); 8,57 с (1Н, HAr); 10,11 ш. с (2Н, N + H2). Основание (9е) также может быть получено по методике, описанной для соединения (9д).
2-[N-(Антрацен-9-илметил)-N-(метил)амино]этан-1-ол гидроиодид (10·HI). Растворяли 2,70 г (10 ммоль) амина (4д) в 70 мл сухого ацетона, добавляли 1,3 мл (20 ммоль) метилиодида, тщательно размешивали и оставляли стоять до следующего дня. Затем раствор кипятили в течение 3–4 ч, упаривали до объема 15–20 мл и охлаждали. Выпавший осадок отфильтровывали, промывали охлажденным ацетоном (2×5 мл), сушили и перекристаллизовывали из 2-пропанола или бутанола. Выход 74 %. Т.пл. 229–230 °С (2-пропанол). Спектр ЯМР 1Н, δ, м.д. (DMSO-d6): 2,72 с (3H, J 5,2 Гц, CH3); 3,41–3,68 м (2Н, СН2); 3,86–4,12 м (2Н, СН2); 5,32–5,66 м (3Н, СН2, OH); 7,52–7,76 м (4Н, HAr); 8,15 д (2Н, J 8,8 Гц, HAr); 8,56 д (2Н, J 8,8 Гц, HAr); 8,76 c (1Н, HAr); 10,30 ш. с (1Н, N + H).
Основание (10). Растворяли 10 ммоль гидроиодида (10·HI) в 10 мл ДМФА и добавляли к полученному раствору избыток конц. раствора аммиака (~30–40 мл), тщательно размешивали и через 30 мин выпавший осадок основания (10) отфильтровывали, промывали водой (3×20 мл) и сушили при 60–65 °С в течение 5–6 ч. Выход 83 %.
N-(Антрацен-9-илметил)-N-(2-хлорэтил)-N-метиламин гидрохлорид (11·HCl). Метод А. Получали алкилированием основания (9д) по методике синтеза метильного производного (10·HI) с дальнейшим переводом гидроиодида в гидрохлорид. Выход 82 %. Т.пл. 183–184 °С (бутанол). Спектр ЯМР 1Н, δ, м.д. (DMSO-d6): 2,68 д (3H, CH3); 3,68–3,95 м (2Н, СН2); 4,10–4,30 м (2Н, СН2); 5,36–5,52 м (2Н, СН2); 7,50–7,73 м (4Н, HAr); 8,13 д (2Н, J 8,7 Гц, HAr); 8,62 д (2Н, J 8,7 Гц, HAr); 8,74 c (1Н, HAr); 10,95 ш. с (1Н, N + H).
Метод Б. Основание (10) хлорировали по методике синтеза гидрохлорида (9д·HCl). Выход 89 %. Полученное соединение согласно физико-химическим и спектральным данным идентично полученному по методу А.
Результаты исследования и их обсуждение
Ранее нами было показано, что использование различных галогеналкильных систем, содержащих азотный донорный центр и антраценовый флуорофор в качестве агентов для модификации поли(1-винилимидазола), позволяет создавать эффективные флуорогенные и хромогенные хемосенсорные материалы для определения таких анионов, как F-, AcO- [1].
С целью изучения влияния длины алкильной цепочки, структуры рецепторного узла и заместителей при азотном центре на изменение ионохромных свойств (направленности и селективности) полимерных хемосенсоров синтезирован ряд новых (антрацен-9-илметил)содержащих синтонов.
Разработанные методики получения N-(антрацен-9-илметил)замещенных аминов были распространены на 2-метилпропан-1-амин, S-(-)-1-фенилэтан-1-амин, 2-метоксианилин, 3,5-дихлоранилин, 2-аминоэтанол и 3-аминопропан-1-ол.
Взаимодействием данных аминов (2а-е) с антрацен-9-карбальдегидом (1) была синтезирована серия азометинов (3а-е), которые подвергались восстановлению с применением NaBH4 (схема 1). Восстановление сопровождается появлением в ЯМР 1Н спектрах сигналов протонов метиленовой (непосредственно связанной с антраценовым фрагментом) и NH-группы, а также исчезновением сигнала CH = -фрагмента.
Полученные дизамещенные амины (4а-е) были подвергнуты дальнейшей модификации. Так, взаимодействием соединений (4а-г) с хлорангидридом хлоруксусной кислоты (5) получались хлорацетамиды (6а-г) (схема 2).
Согласно данным ЯМР 1Н спектроскопии ацилирование приводит к исчезновению сигналов NH-групп и появлению синглета протонов CH2Cl-фрагмента.
Для изучения влияния расстояния между полимерной основой и ионактивным фрагментом на хемосенсорные свойства амидных производных реакцией аминов (4б, г) с 3-хлорпропионилхлоридом (7) были синтезированы 3-хлорпропиониламиды (8б, г).
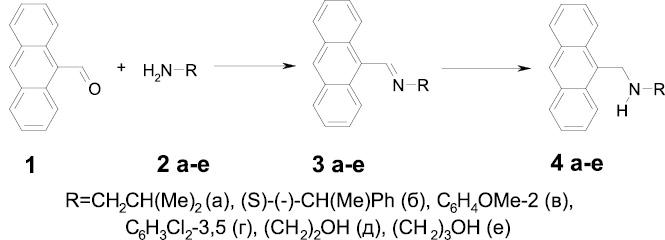
Схема 1
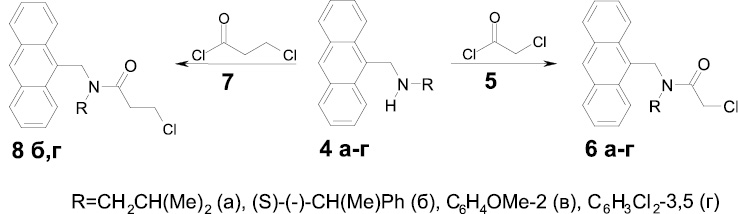
Схема 2
В ходе проведенных исследований разработаны методики синтеза оригинальных алкилхлоридов (8д, е, 11), которые являются важными синтономи при выполнении работ по направленной модификации полимеров. Полученные ранее спирты (4д, е) подвергались хлорированию под действием избытка хлористого тионила при кипячении в хлороформе в течение 3–4 ч. Конечные продукты выпадают из реакционной массы в виде гидрохлоридов. Метильное производное (11) было синтезировано как непосредственным алкилированием хлорэтиламина (9д), так и из аминоспирта (10) (с последующим хлорированием) (схема 3). В качестве растворителей для хлорирования также могут быть использованы безводные бензол или толуол.
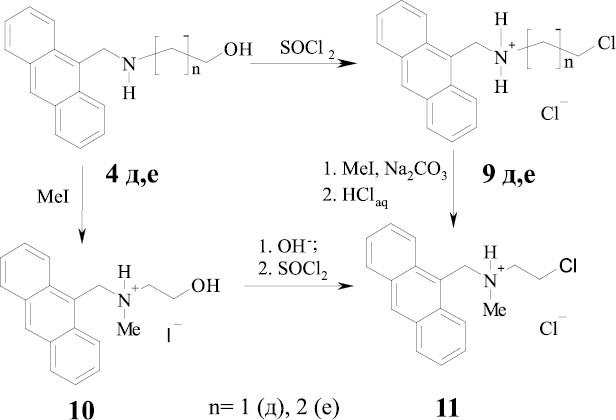
Схема 3
Таким образом, разработаны методы синтеза, получен и охарактеризован ряд ценных прекурсоров – N-(антрацен-9-ил)производных амидов (ацетамидов и 3-пропионамидов) и ω-хлоралкиламинов.
Работа выполнена при финансовой поддержке Минобрнауки России в рамках ФЦП «Исследования и разработки по приоритетным направлениям развития научно-технологического комплекса России на 2007–2013 годы» (проект № 14.A18.21.0803).
Рецензенты:
Стариков А.Г., д.х.н., ведущий научный сотрудник ЮНЦ РАН, г. Ростов-на-Дону;
Михайлов И.Е., д.х.н., профессор, зав. отделом ЮНЦ РАН, г. Ростов-на-Дону.
Работа поступила в редакцию 28.11.2012.
Библиографическая ссылка
Федянина А.Ю., Толпыгин И.Е., Старикова А.А., Николаева О.Г., Левитина И.В., Цуканов А.В., Дубоносов А.Д., Брень В.А. НОВЫЕ ПУТИ СОЗДАНИЯ ХЕМОСЕНСОРНЫХ МАТЕРИАЛОВ // Фундаментальные исследования. 2012. № 11-6. С. 1526-1530;URL: https://fundamental-research.ru/en/article/view?id=30833 (дата обращения: 06.08.2026).