



Если загрязняющие вещества легко окисляются, как пары углеводородов в отходящих газах растворителей и красок, то их удаление может быть осуществлено путем сжигания газов, причем образуется CO2 и H2O. Если концентрация этих примесей в газах достаточно велика и входит в область воспламеняемости, после первоначального поджигания будет поддерживаться процесс самоокисления.
Температура, выше которой горение газов или паров поддерживается произвольно, называется температурой самовоспламенения. Условия для подобного тепловыделения создают за счет сжигания кислорода в количестве 10 - 15 %, превышающем стехиометрическое соотношение, хорошего перемешивания реагентов и оптимального времени их перемешивания в зоне горения. Эти факторы определяются конструкцией горелки и камеры сжигания, а также степенью предварительного смешения газов [1].
Основная химическая реакция окисления любого углеводорода CmHn имеет вид:
CmHn + (m + n/4) O2 → m CO + 2/n H2O + ΔH. (1)
где: O2 - молекулярный кислород, СО - оксид углерода, m и n - целые числа. Выделяющееся тепло ΔH является теплотой реакции (1). Согласно второму закону термодинамики:
ΔG = ΔH - T ΔS, (2)
где: ΔG - изменение энергии Гиббса,Δ S - изменение энтропии; T - абсолютная температура. При отрицательных значениях ΔG реакция возможна, при ΔG > 42 •103 Дж/ кмоль самопроизвольная реакция горения невозможна, в области 0 < ΔG< 42 •103 Дж/ кмоль ее вероятность невелика. Константа равновесия реакции Kp:
Kp = 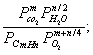 (3)
(3)
где: P- парциальное давление веществ. Существует ряд конструктивных решений, дающих более или менее равномерное перемешивание компонентов и требуемое время пребывания в камере сжигания [2]. К таким камерам сгорания относятся горелки с отталкивающим распределениям пламени (2-3). Температура при этом ограничена 4500 С, поэтому в таких камерах возможна лишь частичная конверсия углеводородов.
В горелках с тангенциальной подачей газов газы входят в камеру сгорания через отражатель пламени, который способствует концентрированию газов у стенок. Этот тип камер сгорания пригоден в тех случаях, когда концентрация кислорода в сжигаемых газах мала, а объемная скорость газов изменяется в широких пределах. В камерах поддерживается температура в пределах 480 - 760 0 С.
В диапазоне температур 250-4000С каталитическое горение происходит с технологически приемлемой скоростью [3]. Современные качественные катализаторы позволяют осуществлять сжигание при 2500С. При 3000С достигается 90% эффективность процесса, а при 350 - 4000С эта величина составляет 99%. Однако возможен кратковременный подъем температуры при 10000С без разрушения катализатора [4]. Термокаталитический метод в результате снижения температуры процесса в 2 - 2,3 раза дешевле по сравнению с термическим сжиганием. Этот метод применяется для очистки газов, содержащих CO, органические соединения, и обеспечивает более полное удаление примесей, чем термическое сжигание[5].
Брутто - схема этого для газовой реакции A + B = C в присутствии катализатора K может быть представлена следующим образом:
A + B + K → K[AB], K[AB] → C + K, (4)
где: K[AB] - активированное промежуточное соединение на поверхности катализатора. Скорость каталитических реакций выражают уравнением w:
w = k • C1a • C2b, (5)
где: C1, C2 - концентрации веществ, участвующих в реакции; k -константа скорости лимитирующей химической реакции; a, b - порядки реакции по соответствующему компоненту. Изменение реакционного пути химического взаимодействия в присутствии катализатора в соответствии с указанным выше механизмом приводит к понижению его энергии активации, что и следует из уравнения Аррениуса:
k = k0 • exp( - E/RT), (6)
где: k0 - предэкспоненциальный множитель; E - энергия активации; R - газовая постоянная.
В некоторых типах каталитических взаимодействий с понижением энергии активации понижается k0. Поэтому рассчитанное на основании снижения энергии активации увеличение константы скорости и соответственно скорости каталитического взаимодействия несколько превышает действительное (4-6). В случае, когда k0 не изменяется, ускоряющее действие катализатора выражают его активностью A. Активность катализатора обычно определяется совокупностью физико-химических свойств, как самого катализатора, так и конвертируемого газового потока. В наибольшей степени она зависит от температуры каталитического превращения, структуры катализатора, содержания в нем промоторов, давления, объемного расхода и продуктов конверсии в газовой смеси.
Активность катализатора может быть выражена количеством продукта (Gп), получаемого с единицы объема (V) катализатора:
A = Gn / ( ![]() •V), (7)
•V), (7)
где: ![]() - время пребывания. Каталитические процессы могут замедляться образующимися продуктами реакции. В этом случае в уравнение скорости реакции следует вводить значения концентраций продуктов реакции. Торможение каталитических процессов продуктами реакции может объясняться двумя причинами (7). Одной из них является адсорбция продуктов реакции поверхностью катализатора, что уменьшает его рабочую часть.
- время пребывания. Каталитические процессы могут замедляться образующимися продуктами реакции. В этом случае в уравнение скорости реакции следует вводить значения концентраций продуктов реакции. Торможение каталитических процессов продуктами реакции может объясняться двумя причинами (7). Одной из них является адсорбция продуктов реакции поверхностью катализатора, что уменьшает его рабочую часть.
В кинетической области скорость процесса определяется только скоростью химического превращения, а в диффузионных областях лимитируется скоростью подвода реагирующих веществ к внешней или внутренней поверхности катализатора и соответственно скоростью отвода продуктов реакции. При очистке других газов реакция протекает в диффузионных областях. Количественно скорость реакции, протекающей во внешнедиффузионной области, определяется законом Фика W:
W = - D • F • ![]() • dc/dn, (8)
• dc/dn, (8)
где: D - молекулярный коэффициент диффузии, F - единичная поверхность катализатора, c - концентрация реагента. Для уменьшения внешнедиффузионного торможения необходимо увеличить линейную скорость газового потока через слой катализатора (8). Явление внутридиффузионного торможения связано с пористой структурой катализатора. Следует различать два типа диффузии: молекулярную и кнудсеновскую.
Первый тип диффузии характерен для крупных пор, размеры которых превышают среднюю длину свободного пробега молекул. Кнудсеновская диффузия протекает в тонких порах. Особое значение этот тип диффузии имеет для процессов, происходящих при низких давлениях.
При сопоставлении скоростей химического превращения и диффузии реагирующих веществ внутри зерен катализатора было сделано заключение о пористой оптимальной структуре катализатора. Для быстрых реакций, когда скорость химического превращения в глубине зерен заметно ниже, чем на их внешней поверхности, наиболее выгодны катализаторы с размером пор, близким к средней длине свободного пробега реагирующих молекул.
Особенно эффективен катализатор неоднородной структуры с крупными порами, превышающими длину свободного пробега, к стенкам которых примыкают короткие тонкие капилляры, отличающиеся большой поверхностью. Явление внутридиффузионного торможения приводит к тому, что не весь объем зерна катализатора участвует в реакции. В условиях промышленной эксплуатации катализатора чащ всего используется не более 20-40% объема катализатора. Из этого следует, что для снижения влияния внутридиффузионного торможения необходимо увеличивать геометрическую поверхность катализаторов.
СПИСОК ЛИТЕРАТУРЫ:
- Володин Н.И., Соколов Э. М. Очистка газовых выбросов. Т.: Химия, 1999,259 с.
- Страус В. Промышленная очистка газов. М.: Химия,1987, 616 с.
- Бретшнайдер Б., Курфюст И. Охрана воздушного бассейна от загрязнений. Л.: Химия,1989, 288 с.
- Чикина Г.А., Мягкий О.Н. Ионообменные методы очистки веществ. В.: Химия, 1984,372 с.
- Попова Н. М. Катализаторы очистки газовых выбросов промышленных производств. М.: Химия, 1991, 175 с.