


Scientific journal
Fundamental research
ISSN 1812-7339
"Перечень" ВАК
ИФ РИНЦ = 1,798
NEW STRATEGY OF IMMUNE RESPONSE MANAGEMENT IN PULMONARY DISEASES ‒ ROLE OF SURFACTANT PROTEIN D AS BIVALENT MACROPHAGES REPROGRAMMING FACTOR

По данным Всемирной организации здравоохранения (ВОЗ), в России и мире заболевания легких с воспалительным компонентом ‒ хроническая обструктивная болезнь легких (ХОБЛ), бронхиальная астма (БА), болезни органов дыхания инфекционной этиологии, саркоидоз органов дыха-
ния ‒ прочно занимают лидирующие позиции в структуре общей заболеваемости и смертности трудоспособного населения. Данные заболевания вносят существенный вклад в рост нетрудоспособности, увеличение инвалидности и преждевременной смертности населения [1].
Иммунный ответ при заболеваниях легких
Основным звеном патогенеза заболеваний легких является дисбаланс клеточного Th1 и гуморального Th2 звеньев иммунного ответа, обусловливающий «неадекватное течение» воспалительной реакции [1, 4]. В связи с этим понимание молекулярных и клеточных механизмов воспаления в легких и возможности их коррекции является одной из фундаментальных проблем медицины и представляет собой одно из значимых и перспективных направлений современных клинических исследований.
Исключительно важную роль в иммунных ответах играют макрофаги. Это связано с тем, что внедрение в организм чужеродного агента вызывает прежде всего мощную активацию макрофагов, выделение цитокинов и других медиаторов воспаления. При этом в зависимости от микроокружения макрофаги могут существенно модифицировать свою активность и выполнять роль про- и антивоспалительных агентов. При наличии в среде низких доз бактериального липополисахарида (ЛПС) и/или IFN-γ макрофаги в ответ на действие стимулирующих факторов отвечают классической активацией, т.е. продукцией цитокинов провоспалительного профиля, таких, как IL-1, TNF-α, IL-12 и IFN-γ, а также генерацией активных форм кислорода и азота. Такой фенотип макрофагов получил название M1 [2; 13]. Для M1 макрофагов характерны выраженные фагоцитирующие и бактерицидные свойства и способность к уничтожению внутриклеточных микробов, таких, как вирусы и бактерии, а также опухолевых клеток. При наличии в среде таких факторов, как IL-4, IL-13, TGF-ß или глюкокортикоиды, макрофаги формируют альтернативный М2 фенотип, продуцирующий антивоспалительные цитокины, такие, как IL-10, IL-13, в ответ на действие стимулирующих факторов [2; 13]. M2 макрофаги способны к уничтожению экстраклеточных паразитов, таких, как гельминты и грибы, содействуют ангиогенезу, репарации и ремоделированию тканей, но при этом могут способствовать и опухолевому росту. Для подчеркивания контраста с «классической активацией» М1 макрофагов особая активация М2 фенотипа обозначается новым термином «альтернативная активация».
Показано, что макрофаги М1 фенотипа интегрированы в Th1 клеточный иммунный ответ, а макрофаги М2 фенотипа ‒ в Th2 гуморальный иммунный ответ; набор цитокинов, продуцируемых М1 макрофагами, аналогичен цитокинам, продуцируемым Th1 клетками, а М2 макрофагами - Th2 клетками [14].
Общепризнано, что в определенной степени состояние иммунного статуса организма, и прежде всего сбалансированность иммунного ответа Th1/Th2, обусловливают течение и прогноз заболеваний, а также развитие осложнений с присутствием воспалительного компонента [14]. «Неадекватная» активация Th1 или Th2 звена иммунного ответа и, соответственно, изменение необходимого баланса Th1/Th2 ответов может способствовать и лежать в основе различных заболеваний, в том числе и бронхо-легочной системы [14].
При развитии заболеваний легких с воспалительным компонентом отмечается дисбаланс в системе Th1/Th2 ответов, что позволяет предположить и изменение процесса репрограммирования макрофагов на М1 или М2 фенотип. При этом альвеолярные макрофаги выделяют медиаторы воспаления, способствующие развитию бронхиальной обструкции вследствие отека, гиперсекреции слизи, изменению реологических свойств мокроты, морфологической перестройке бронхиального дерева и дальнейшей колонизации бронхиального дерева патогенными микроорганизмами.
В связи с этим крайне важно установление и достижение определенного баланса М1/М2 для формирования необходимого «терапевтического» соотношения Th1/Th2 иммунного ответа при той или иной патологии легких, что может обеспечить эффективное выздоровление и предотвращение обострения хронических заболеваний легких у пациентов.
Сурфактантный белок D и его роль в иммунном ответе легких
В ходе проведенных исследований было показано, что компонент сурфактанта легких ‒ сурфактантный белок D (SP-D) является уникальным фактором репрограммирования, действующим по принципу «два в одном», т.е. способным репрограммировать макрофаги и на М1, и на М2 фенотип [7]. SP-D ‒ представитель семейства коллагеноподобных лектинов (коллектинов), играющих значимую роль во врожденном иммунитете и антителонезависимом иммунном ответе [7]. SP-D ‒ компонент сурфактанта легких, продуцируемый альвеолярными клетками II типа и нецилиарными клетками бронхиол легких ‒ клетками Клара [12]. Основная функция SP-D заключается в модулировании иммунной защиты и воспаления. Понимание важности SP-D для иммунной защиты легких в целом и адекватного функционирования альвеолярных макрофагов, в частности, возникло после экспериментов на мышах, геном которых не имел гена SP-D (SP-D (-/-)). Показано, что отсутствие гена SP-D у экспериментальных мышей приводит к значительному увеличению воспалительных реакций в легких [15; 16], и в частности, увеличению уровня провоспалительных цитокинов, таких, как IL-6 и IL-12, прогрессивному развитию фиброза в субплевральных зонах [6; 17] и, в конечном итоге, развитию эмфиземы легких [6]. Кроме того, у SP-D (-/-) мышей отмечалось снижение массы тела, и возрастала восприимчивость к инфекциям [11]. Установлено, что SP-D стимулирует хемотаксис нейтрофилов и связывается с альвеолярными макрофагами in vitro, а также участвует в усилении захвата микробов макрофагами [7].
В легких SP-D (-/-) мышей также увеличивалось содержание макрофагов, находящихся на разных стадиях некроза и апоптоза. Важно, что добавление экзогенного SP-D ограничивало гибель макрофагов. При этом SP-D за счет связывания с углеводными и липидными частями на поверхности апоптотических клеток облегчал процесс фагоцитоза уже погибших клеток и, таким образом, способствовал нормальному разрешению воспаления [8].
Результаты проведенных исследований показали, что SP-D может существовать в различных олигомерных состояниях ‒ в форме мономера, тримера, додекамера или мультимера [5; 9]. Мономер белка SP-D имеет молекулярную массу 43 кДа, состоит из 375 аминокислот и включает четыре домена: NH2-хвостовой домен, коллагеноподобный домен, домен «шейки» и С-концевой лектиновый домен «головка», распознающий COOH- группы углеводов и лектин С-типа [16]. В состав зрелой формы белка SP-D входит 375 аминокислот (рис. 1).

Рис. 1. Доменная структура SP-D
Белок SP-D содержит 7 цистеиновых остатков, 2 из которых могут быть мишенью для S-нитрозилирования (в положении 15 и 20), что является важной структурной особенностью SP-D [5]. SP-D мономеры (43 кDa) могут объединяться в тримеры. Эти тримеры могут объединяться в тетрамеры, формируя додекамер [5] (рис. 2).
![Возможные олигомерные формы существования SP-D [5]](/fs/i/2011/1/10.jpg)
Рис. 2. Возможные олигомерные формы существования SP-D [5]
Для процесса олигомеризации SP-D важно наличие цистеинов в положении 15 и 20 [18]. Вовлечение цистеиновых остатков, подвергающихся S-нитрозилированию, предполагает участие NO в контролировании олигомеризационного состояния SP-D. Современные исследования показали, что олигомеризация SP-D является ключевым моментом в регуляции воспалительного ответа в легких [18]. Кроме того, было установлено, что разные олигомерные формы SP-D альтернативно влияют на активность и функции альвеолярных макрофагов [9]. Это связано с тем, что мультимеры и додекамеры SP-D взаимодействуют с одним типом рецепторов на поверхности альвеолярных макрофагов, тогда как S-нитрозилированные тримеры - с другим типом рецепторов [9]. Позже группа исследователей под руководством А. Gow (2004) доказала, что NO за счет образования SNO в положении «цистеин 15» может регулировать образование додекамеров SP-D и, таким образом, представлять сигнальный механизм в переключении роли SP-D с ингибитора воспалительной активности в активатор (рис. 3).
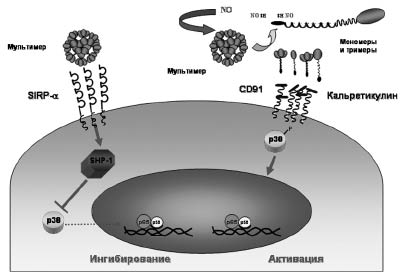
Рис. 3. Схема про-и противовоспалительных функций сурфактантного белка D (SP-D) (модифицированная схема Gardai, 2003) [9]
SP-D, являясь компонентом микроокружения макрофагов в легких, представляет собой также и один из ключевых регуляторов их активности. Показано, что SP-D не только регулирует баланс макрофагальных цитокинов Th1 и Th2 профиля [3], но и вовлечен в регуляцию стресс-ответа макрофагов [2]. При анализе продукции цитокинов у мышей, имеющих ген SP-D (SP-D(+/+)) и лишенных гена SP-D (SP-D(-/-)), установлено, что SP-D фенотипзависимо регулирует баланс макрофагальных цитокинов Th1 и Th2 профиля. Показано, что зависимость влияния SP-D на продукцию разных Th1 и Th2 цитокинов от фенотипа макрофагов предопределяет изменение баланса Th1/Th2 цитокинов. Так, в ЛПС-стимулированном нативном М0 фенотипе SP-D (-/-) макрофагов за счет снижения продукции Th2 цитокинов IL-10 и IL-13 Th1/Th2 баланс цитокинов сдвигается в сторону Th1 цитокинов. В М1 фенотипе отсутствие SP-D оказывает разнонаправленный эффект на разные Th1 и Th2 цитокины. В М2 фенотипе за счет усиления продукции продукции Th2 цитокинов баланс Th1/Th2 цитокинов сдвигается в сторону Th2 [3].
Представленные данные позволяют предположить, что SP-D играет роль эндогенного фактора репрограммирования макрофагов. К настоящему времени обнаружены факторы, под воздействием которых возможно репрограммирование макрофагов на М1 фенотип. Установлено, что к ним относятся Тh1 цитокины (IFN-γ, ФНО-α), патоген-ассоциированные молекулярные комплексы ‒ ЛПС, липопротеины, различные грамположительные и грамотрицательные микроорганизмы, цитомегаловирус, белки теплового шока, компоненты внеклеточного матрикса [10; 12; 13]. Установлены также факторы, действие которых программирует макрофаги на М2 фенотип: это Тh2 цитокины (IL-4, IL-13), иммунокомплексы в сочетании с IL-1β, IL-10, TGF-β, в некоторых ситуациях ‒ внутриклеточные патогены (Coxiella burnetii, Leismania), витамин D3, гормоны (глюкокортикоиды), апоптотические клетки [10; 12; 13]. Но лишь SP-D ‒ уникальный эндогенный фактор, действующий по принципу «два в одном», т.е., способный в зависимости от своей олигомерной структуры репрограммировать макрофаги как на М1, так и на М2 фенотип.
Таким образом, возможность воздействия на патогенетические звенья воспалительной реакции с помощью SP-D-зависимого репрограммирования макрофагов и достижения необходимой сбалансированности Th1/Th2 ответов можно рассматривать как новую стратегию управления иммунным ответом при заболеваниях с воспалительным компонентом, в том числе и заболеваниях легких.
Работа выполнена в рамках реализации ФЦП «Научные и научно-педагогические кадры инновационной России» на 2009-2013 годы.
Список литературы
- Стандарты по диагностике и лечению больных хронической обструктивной болезнью легких (ATS/ERS, пересмотр 2004 г.) / под ред. А.Г. Чучалина; пер. с англ. ‒ М.: Атмосфера, 2005. ‒ 96 с.
- Вассерман Е.Н., Абрамова Е.В., Круглов С.В. и соавт. Отсутствие гена SP-D приводит к усилению ЛПС-индуцированного синтеза HSP70 в М2, но не в М1 фенотипе перитонеальных макрофагов: возможная роль интерлейкина-10 // Фундаментальные исследования. ‒ 2010. ‒ №6. ‒ С. 19-27.
- Вассерман Е.Н., Лямина С.В., Шимшелашвили Ш.Л. и соавт. SP-D контролирует баланс Th1 и Th2 цитокинов и обладает признаками эндогенного фактора репрограммирования макрофагов// Фундаментальные исследования. ‒ 2010. ‒ №6. ‒ С. 28-36.
- Хаитов Р.М. Иммунология: учебник. ‒ М.: ГЭОТАР-Медиа, 2006.
- Atochina E.N., Beers M.F., Hawgood S. et al. Surfactant protein-D, a mediator of innate lung immunity, alters the products of nitric oxide metabolism // Am J Respir Cell Mol Biol, 2004; 30: 271-279.
- Botas C.F., Poulain J., Akiyama J. et al. Altered surfactant homeostasis and alveolar type II cell morphology in mice lacking surfactant protein D // Proc. Natl. Acad. Sci, 1998; 11869-11874.
- Crouch E.C. Structure, biologic properties and expression of surfactant protein D // Biochim. Biophys. Acta, 1998; 1408: 278-289.
- Fisher J.H., Larson J., Cool C., Dow S.W. Lymphocyte activation in the lungs of SP-D null mice // Am J Respir Cell Mol Biol, 2002; 27: 24-33.
- Gardai S.J., Xiao Y.-Q., Dickinson M. et al. By binding SIRP-alpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation // Cell, 2003; 115: 13-23.
- Gordon S. The macrophage: Past, present and future // Eur. J. Immunol 2007; 37: S9.
- LeVine A.M., Whitsett J.A., Gwozdz J.A. et al. Distinct effects of surfactant protein A or D deficiency during bacterial infection on the lung // J Immunol 2000; 165: 3934-3940.
- Mantovani A, Sica A, Locati M., Macrophage Polarization Comes of Age // Immunity, 2005; 23(4): 344-346.
- Martinez F.O., Sica A., Mantovani A., Locati M. Macrophage activation and polarization //
Front Biosci., 2008; 1(13): 453-61. - Oosterhout A.J.M., Motta A.C. Th1/Th2 paradigm: not seeing the forest for the trees? // Eur Respir J, 2005; 25: 591-593.
- Sin D.D., Pahlavan P.S., Man. P.S. Paul. Surfactant Protein D: A Lung Specific Biomarker in COPD?: Potential Biological Roles of SP-D in COPD // Ther Adv Resp Dis 2008; 2(2): 65-74.
- Sorensen G.L., Husby S., Holmskov U.. Surfactant protein A and surfactant protein D variation in pulmonary disease // Immunobiology, 2007; 212 (4-5): 381-416.
- Wert S.E., Yoshida M., LeVine A.M. et al. Increased metalloproteinase activity, oxidant production, and emphysema in surfactant protein D gene-inactivated mice // Proc. Nat. Acad. Sci 1997; 7: 5972-5977.
- Zhang L., Ikegami M., Crouch E.C. et al. Activity of pulmonary surfactant protein-D (SP-D) in vivo is dependent on oligomeric structure // J. Biol. Chem. 2001; 276 (22): 19214-19219.
- Zhang X., Morrison D.C. Lipopolysaccharide-induced selective priming effects on tumor necrosis factor α and nitric oxide production in mouse peritoneal macrophages // J. Exp. Med, 1993; 177(2): 511-516.
Рецензент:
Пшенникова М.Г., д.б.н., профессор, старший научный сотрудник ГУ НИИ общей патологии и патофизиологии РАМН.
Библиографическая ссылка
Лямина С.В, Круглов С.В, Веденикин Т.Ю, Малышев И.Ю НОВАЯ СТРАТЕГИЯ УПРАВЛЕНИЯ ИММУННЫМ ОТВЕТОМ ПРИ ЗАБОЛЕВАНИЯХ ЛЕГКИХ ‒ РОЛЬ СУРФАКТАНТНОГО БЕЛКА D КАК БИВАЛЕНТНОГО ФАКТОРА РЕПРОГРАММИРОВАНИЯ МАКРОФАГОВ // Фундаментальные исследования. 2011. № 1. С. 90-98;URL: https://fundamental-research.ru/en/article/view?id=15813 (дата обращения: 04.07.2026).