



На современном этапе развития геномики человека использование биоинформатических методов является необходимым для интерпретации полученных данных, поиска генов-кандидатов для анализа геномных ассоциаций, определения молекулярных механизмов заболеваний, увеличения эффективности сканирования генома [2–6, 10, 12–15, 18, 20, 23]. Более того, при изучении возможных фенотипических последствий генетической патологии необходима крайне строгая оценка последствий изменения последовательностей ДНК на молекулярном, супрамолекулярном и клеточном уровнях [6, 12, 15, 17, 23]. Одним из биоинформатических методов, позволяющих идентификацию патогенности геномных перестроек и ассоциации вариабельности последовательности ДНК с определенными фенотипическими признаками, является приоритезация генов-кандидатов [4, 17]. Поскольку функциональные характеристики генов являются относительно постоянными параметрами, совокупность данных относительно изменений их числа копий может быть использована для оценки последствий генных и хромосомных мутаций [1, 7, 9, 12, 15–19]. Тем не менее при молекулярно-цитогенетическом анализе геномных и хромосомных аномалий, являющиеся неотъемлемой частью фундаментальных и диагностических исследований в области медицинской генетики [1, 3, 21, 23, 24], методы in silico используются крайне редко. Это, по-видимому, связано с тем, что успешное применение биоинформатических технологий сопряжено с необходимостью глубоких познаний в области информатики, математической статистики и других подобных дисциплин. С другой стороны, для изучения влияния вариаций числа копий последовательностей ДНК (CNV), выявляющихся у каждого индивидуума с помощью молекулярно-цитогенетических методов сканирования генома, на фенотип необходимо обладать технологиями, которые позволяют определить их патогенность без расширенной панели дополнительных исследований [2, 9, 12, 15, 23]. Следовательно, задача разработки биоинформатических технологий для оценки функциональных последствий геномных вариаций является актуальной для современной биомедицины.
В последнее время были предложены методы изучения генома in silico, послужившие основой для разработки более сложных технологий выявления хромосомных перестроек и обобщения геномных данных [5, 6, 12–19]. В настоящей работе эти методы были использованы для создания технологии определения последствий геномных вариаций с помощью идентификации генов и процессов-кандидатов на основе in silico анализа эпигеномных, протеомных и метаболомных баз данных. Было показано, что предлагаемая технология может быть с успехом использована для фундаментальных и прикладных исследований генома с помощью молекулярно-цитогенетических методов.
Материалы и методы исследования
Мы проанализировали результаты исследований 225 пациентов методом серийной сравнительной геномной гибридизации (array CGH) в соответствии с описанными ранее протоколами [12, 14, 15]. Кроме того, были биоинформатически оценены данные анализа более 7000000 клеток из 250 образцов, исследованных методами интерфазной и метафазной многоцветовой FISH. Результаты FISH с использованием сайт-специфических и центромерных ДНК проб были описаны в наших предыдущих работах [1, 7–12, 21, 24].
Геномные, эпигеномные, протеомные и метаболомные данные были проанализированы, как описано ранее [4, 5, 10, 12, 15, 16, 19]. Данные о каждом гене, вовлеченном в хромосомные аномалии и/или CNV, были получены из клинических, геномных (браузеры и базы данных онтологии генов), эпигенетических (экспрессия генов), протеомных, интерактомных (базы данных + программное обеспечение) и метаболомных баз данных. Гены-кандидаты были выбраны в соответствии с данными межтканевой вариабельности экспрессии гена, полученных с помощью ресурса BioGPS [22]. Для подтверждения выбора были использованы протеомные и метаболомные данные. В качестве критерия отбора были предложены эпигенетические (экспрессия) и метаболомные данные. Интерактомный анализ был осуществлен с помощью программного обеспечения и баз данных, описанных ранее [15, 16]. Метаболомные данные были получены из различных источников (баз данных онтологии генов и реактомного анализа [5, 19]).
Приоритезация была осуществлена на основе изучения онтологии генов (т.е. отбора генов в соответствии с их непосредственным отношением к фенотипу или их участия в молекулярных/клеточных процессах, имеющих отношение к признаку). Затем было использовано специфическое ранжирование генов в соответствии с их функциональной нагрузкой. Для завершения приоритезации был использован анализ протеомных сетей, относящихся к мутированным или делетированным/дуплицированным генам). Далее осуществлялась идентификация процессов-кандидатов патологии в каждом конкретном случае. Приоритезация одного или нескольких генов в CNV рассматривалась в качестве основного критерия для подтверждения патогенности соответствующего изменения копийности последовательностей ДНК. Информация из клинических и молекулярных баз данных была также оценена с помощью библиографического анализа уровня публикаций (цитируемость и наличие сходящихся результатов в нескольких публикациях) с помощью ресурсов Google Scholar (https://scholar.google.com) и PubMed (http://www.ncbi.nlm.nih.gov/pubmed).
Результаты исследования и их обсуждение
Вариабельность экспрессии между генами является относительно стабильным параметром. Более того, ряд эпигенетических баз данных (например, BioGPS [22]) предоставляет сведения относительно межтканевой изменчивости уровня экспрессии генов, которая может быть использована в качестве критерия приоритезации [15]. Используя концепцию, предложенную ранее для тканеспецифичной геномной патологии, в частности, сформулированной для нервных и психических заболеваний (основная причина заболеваний головного мозга – геномные изменения, затрагивающие ткань головного мозга) [7–11, 20, 21, 25], можно было заключить, что мутации гена (или CNV/хромосомные перестройки), скорее всего, реализуются за счет патологических изменений в определенной ткани. Последнее, по-видимому, достигается за счет неравномерного распределения экспрессии генов в различных тканях. Следовательно, становится возможным отнести гены, вовлеченные в хромосомные перестройки или CNV, к патологическим клеточным процессам в определенных тканях.
Экспрессия генов рассматривается в качестве определяющего критерия классификации клеточных или патологических состояний и приоритезации геномных вариаций [1, 15, 18, 22]. Вариабельность экспрессии генов может также указывать на изменения в геномных сетях [17, 18]. Для повышения эффективности приоритезации генов при выборе стратегии «фильтрования» и «ранжирования» генов (белков), вовлеченных в геномную патологию [17], также необходимы дополнительные параметры. После выбора генов в соответствии с их профилями экспрессии из протеомных баз данных была получена дополнительная информация (последствия генных мутаций на протеомном уровне – межбелковые взаимодействия или интерактомные сети), геномные сети («реактом») и метаболические процессы (метаболом). Вариации генома были также исследованы на предмет соответствия с непатогенными вариациями последовательностями ДНК (Database of Genomic Variants, http://dgvbeta.tcag.ca). Совокупность полученных данных исследовалась по схеме, приведенной на рис. 1.
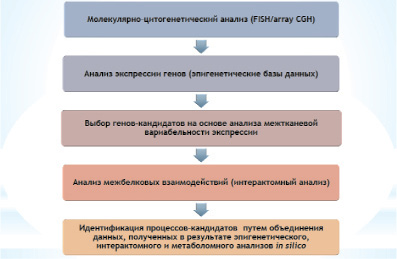
Рис. 1. Блок-схема последовательности осуществления процедур биоинформатического анализа функциональных последствий геномных вариаций: (I) гены, участвующие в геномной перестройке, выбираются в соответствии с профилем экспрессии генов; (II) полученные данные используются для построения геномных сетей; (III) другие элементы (гены) сети оцениваются таким же способом
Благодаря интерактомному анализу применение технологии не ограничивается генами, участвующими в хромосомном дисбалансе или CNV. Результаты биоинформатического анализа могут также быть подтверждены «клинической проверкой» или, другими словами, корреляциями генотип-фенотип.
Структурные хромосомные перестройки и анеуплоидии исследовались с помощью биоинформатической технологии, поскольку при подобном исследовании возможен контроль приоритезации генов с помощью корреляций между генотипом и фенотипом, а также сравнения с ранее полученными данными по картированию хромосомных синдромов. В частности, при трисомии хромосомы 21 (синдроме Дауна) исследовались гены-кандидаты, ассоциированные с патологией мозга. С помощью биоинформатического анализа было обнаружено, что гены CXADR, GABPA, APP, TIAM1, SYNJ1, SON, ATP5O, TTC3, HMGN1, PDXK, SUMO3, S100B и геномные сети, в которых они участвовали, являются генами-кандидатами и процессами-кандидатами соответственно для патологии мозга при трисомии хромосомы 21. Биоинформатический анализ генов-кандидатов, вовлеченных в хромосомные перестройки в когорте пациентов с умственной отсталостью, аутизмом, эпилепсией и/или врожденными пороками развития, показал, что при данных патологических состояниях наблюдается нарушение следующих геномных сетей: репликация и репарация ДНК (включая nucleotide excision/ mismatch repair, DNA damage-ATM-p53-apoptosis pathway, и p53-/MAPK-/ErbB-/PI3KAkt-signaling), регуляция клеточного цикла и сегрегации хромосом в митозе, запрограммированная клеточная гибель (апоптоз), аксональное наведение. Примечательно, что одним из элементов патогенетического каскада при патологии мозга является соматический хромосомный мозаицизм и геномная нестабильность, причиной которых, как правило, являются нарушения вышеуказанных процессов [1, 7–11, 20, 21, 25].
Как правило, первым этапом ассоциации генетических изменений с фенотипическими особенностями является выявление геномных вариаций. При молекулярно-цитогенетическом анализе таковыми являются аномалии хромосом или CNV, которые сужают поиск кандидатов до определенного участка хромосомы или даже гена. Компьютерная аннотация генома при цитогенетическом анализе обеспечивает возможность определения генов, затронутых хромосомными перестройками. Тем не менее подобные ассоциации требуют дополнительных молекулярно-цитогенетических исследований больших когорт и дальнейших исследований предполагаемых генов-кандидатов и их функционального значения на молекулярном и клеточном уровнях. Отсутствие успеха в картировании генов мультифакторных заболеваний свидетельствует о том, что вместо попыток выявления новых генов-кандидатов в качестве мишеней для исследований и терапевтического вмешательства следует рассматривать специфические изменения в молекулярных/клеточных процессах. Таким образом, становится возможным изучение геномных сетей заболеваний на основании молекулярно-цитогенетических данных, что, несомненно, является важным технологическим достижением в области медицинской генетики.
Определение патогенного значения CNV можно по-другому обозначить как приоритезацию CNV. Фенотипические проявления были предложены в качестве основного критерия для осуществления этого процесса. Одним из критериев определения возможных фенотипических последствий данного CNV с помощью настоящей технологии является наличие хотя бы одного приоритетного гена. В общей сложности были приоритезированы 462 CNV у 181 пациента, позволяя оценить диагностический потенциал данной биоинформатической технологии как 88,3 %. Проблема дифференциации патогенных и непатогенных CNV остается до сих пор актуальной [1, 3, 6, 9, 12, 20]. Несмотря на то, что было описано несколько способов решения задачи интерпретации «вариома» (совокупность всех CNV, выявленных у одного пациента или при каком-либо заболевании), до настоящего времени нет общепринятых рекомендаций для оценки патогенности этих форм вариабельности генома. Приоритезация CNV в больших когортах может указать на такие процессы-кандидаты для данного заболевания, как молекулярные и клеточные пути, вовлеченные в сложный каскад биологических процессов (т.е. старение, регуляция клеточного цикла, регуляция транскрипции, ремоделирование хроматина, поддержание стабильности генома). Успешное применение биоинформатического анализа способствует прогрессу в понимании механизмов заболеваний и, как следствие, разработке терапевтических стратегий.
In silico анализ соматических вариаций генома (СВГ) проводился двумя способами: (I) оценка последствий СВГ с точки зрения фенотипа; (II) анализ вариома для поиска причин возникновения СВГ. Первый способ в основном использовался для структурных геномных вариаций и геномной нестабильности (GIN), тогда как последний способ был использован для определения причин происхождения хромосомной нестабильности (CIN) в виде анеуплоидии/полиплоидии. Исследование GIN и CIN, проявляющихся в виде разрывов и перестроек хромосом, позволило выявить локусы генома (ломкие сайты хромосом и хромосомные участки, содержащие повторяющиеся последовательности ДНК) и картирование генов, нарушенных из-за CIN при заболеваниях головного мозга (атаксия-телеангиэктазия и болезнь Альцгеймера) (подробнее см. [10, 25]). Следует отметить, что эти геномные изменения не могут быть обнаружены с помощью высокоразрешающих технологий полногеномного сканирования. Несмотря на то, что такие виды GIN/CIN редко оцениваются с помощью анализа in silico, информация об их воздействии на фенотип может пролить свет на новые генетические механизмы биологического разнообразия и патологии. Было также показано, что немозаичные геномные вариации могут приводить к нарушению регуляции сегрегации хромосом и геномной стабильности, вызывая, в свою очередь, CIN или GIN. Более того, поскольку настоящий метод приоритезации основан на оценке межтканевой вариабельности экспрессии генов, он также может быть использован для изучения тканеспецифичного мозаицизма. Учитывая полученные данные, был сделан вывод о том, что предлагаемая биоинформатическая технология дает возможность определения механизмов и последствий соматического мозаицизма.
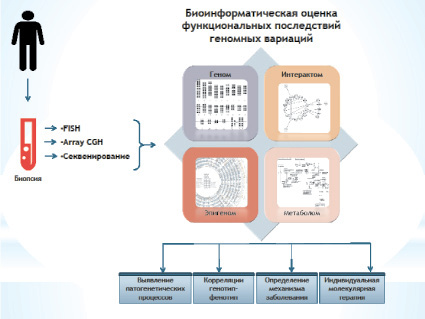
Рис. 2. Блок-схема предлагаемой биоинформатической технологии
Заключение
Использование биоинформатических технологий для оценки результатов анализа генома требует глубоких знаний в области компьютерных наук, статистики и смежных дисциплин. Тем не менее наличие доступных для широкого круга пользователей онлайн-инструментов и программного обеспечения может упростить использование биоинформатических методов. В настоящей работе мы предложили технологию приоритезации генов-кандидатов и CNV на основе анализа геномных, эпигеномных, протеомных и метаболомных баз данных/онлайн-инструментов. Преимуществом этой биоинформатической технологии является универсальность, или, точнее, отсутствие необходимости использования специального программного обеспечения или баз данных. Молекулярно-цитогенетические методы полногеномного сканирования и хромосомного анализа соматических клеток в сочетании с биоинформатической оценкой транскриптомных и протеомных данных позволяют определить возможную причинно-следственную связь между соматическим мозаицизмом и немозаичными хромосомными/геномными перестройками. Это может способствовать определению корреляций генотип-фенотип и механизмов болезни, а также развитию персонализированной молекулярной терапии. Полученные данные создают основу для нового направления в биомедицине – молекулярной цитогенетики in silico, основные принципы которой можно изобразить с помощью блок-схемы предлагаемой биоинформатической технологии (рис. 2).
Выводы
Для проведения in silico оценки молекулярно-цитогенетических данных, мы предложили биоинформатическую технологию оценки функциональных последствий геномных вариаций с помощью приоритезации генов-кандидатов и CNV. Данный комплекс методов обладает следующими преимуществами: (I) универсальность, достигаемая за счет отсутствия необходимости использования определенных баз данных и программного обеспечения; (II) относительная простота алгоритма (возможность избежать сложных вычислений и дополнительного статистического анализа); (III) принятие во внимание специфики данных, полученных молекулярно-цитогенетическими методами. Таким образом, небезосновательно утверждение о том, что предлагаемая технология может быть с успехом использована в ходе фундаментальных и диагностических молекулярно-цитогенетических исследований.
Исследование выполнено за счет гранта Российского научного фонда (проект № 14-15-00411).
Библиографическая ссылка
Юров И.Ю., Ворсанова С.Г., Зеленова М.А., Васин К.С., Юров Ю.Б. БИОИНФОРМАТИЧЕСКАЯ ТЕХНОЛОГИЯ ОЦЕНКИ ФУНКЦИОНАЛЬНЫХ ПОСЛЕДСТВИЙ ГЕНОМНЫХ ВАРИАЦИЙ // Фундаментальные исследования. 2015. № 2-19. С. 4209-4214;URL: https://fundamental-research.ru/ru/article/view?id=37931 (дата обращения: 30.06.2026).