



Заболевания сердечно-сосудистой системы занимают одно из первых мест, а летальность от них составляет более 50 % от общей смертности населения [3]. За последнее десятилетие этот показатель снижается, но все же остается достаточно высоким. Указанные заболевания, как правило, сопровождаются аритмиями, которые могут вызывать различные факторы. Большинство антиаритмических препаратов, традиционно применяющихся при лечении указанной патологии, обладают недостаточным лечебным эффектом при монотерапии, а использование некоторых из них сопряжено с появлением нежелательных реакций. Это подчеркивает целесообразность использования кардиостимуляторов, а также применения персонализированного, индивидуального подхода к каждому пациенту, включая необходимость разработки и внедрения в медицинскую практику новых современных антиаритмических лекарственных средств.
Как потенциальное антиаритмическое средство представляет интерес синтезированное в Пермской государственной фармацевтической академии [5] соединение – 2-метиланилид-N,N-диэтиламиноэтановой кислоты нитрат, получившее условное название мономекаин. Скрининговые исследования показали, что мономекаин в сравнении со своим структурным аналогом лидокаином проявляет более выраженное антиаритмическое действие. Его антиаритмический индекс превосходит таковой лидокаина в 9 раз, а токсичность меньше в 1,6 раза [4]. На доклинической стадии испытания соединения необходимы исследования, направленные на разработку способов оценки его качества, стандартизации и последующего создания нормативного документа – фармакопейной статьи (ФС).
Целью настоящего исследования является разработка методики количественного определения мономекаина, т.к. качество субстанции, используемой в доклинических испытаниях, имеет важное значение.
Материалы и методы исследования
Объект исследования – мономекаин – 2-метиланилид-N,N-диэтиламиноэтановой кислоты нитрат. Эксперименты проведены на 10 сериях биологически активного соединения (БАС), полученных в лабораторных условиях по методике, описанной ранее и модифицированной автором [2].
Чистота и однородность полученных образцов подтверждена элементным анализом, по температуре плавления, спектральными характеристиками (ИК и ЯМР 1Н), тонкослойной хроматографией.
Мономекаин представляет собой белый кристаллический порошок. Легко растворим в воде и спирте этиловом 95 %, умеренно растворим в ацетоне.

Спектры ЯМР 1Н зарегистрированы на приборе Bruker Avance 500 (Германия) (500 МГц для 1Н) в DMSO-d6 с SiMe4 в качестве внутреннего стандарта при комнатной температуре. ИК спектры получены на ИК Фурье спектрометре ALPHA-T в виде диска с калия бромидом в соотношении 1:200 мг. Температура плавления соединения определена на приборе SMP3 (Barloworld Scientific). Элементный анализ образцов выполнен на автоматическом CHN анализаторе Perkin Elmer РЕ 2400, серия II. Для ТСХ использовали пластинки Sorbfil (OOO «Имид», Россия).
Выход продукта (6) 78,5 %; Тпл 137,5–138,5 °С; ТСХ:ацетон:аммиак, 9:1; Rf 0,56.
C13H21N3O4 (283,32 г/моль). Найдено (%): С 55,23; H 7,73; N 14,70. Вычислено (%): C 55,11; H 7,47; N 14,83.
Спектр ЯМР 1Н: 9,96 с (1 Н, NH); 9,45 уш.с (1 Н, HNО3); 7,42 д (1 Н, Ar, J = 7,72); 7,27 д (1 Н, Ar, J = 7,46); 7,22 ддд (1 Н, Ar, J = 7,46; 7,46; 1,20); 7,16 ддд (1 Н, Ar, J = 7,44; 7,44; 1,20); 4,15 с (2 Н, СН2); 3,23 д (4 Н, 2× СН2CH3, J = 6,92); 2,23 с (3 Н, СН3); 1,24 д (6 Н, 2× СН2CH3, J = 7,24).
ИК спектр: полосы поглощения при 3240–3110 см–1 обусловлены валентными колебаниями N–H связей; 3030 см–1 – С–Н связи ароматического кольца; 1690 см–1 – «амид-I»; 1537 см–1 – NH и OCN (полоса амид-II). К валентным колебаниям C-N связи относится полоса при 1261 см–1 (полоса амид-III); валентные колебания нитрат-иона проявляются в виде интенсивной полосы 1384 см–1 и более слабых при 1040 и 825 см–1.
Использованы титрованные растворы, реактивы, растворители, индикаторы, соответствующие требованиям ГФ XII изд. [5]. Потенциометрическое титрование проводили с помощью автоматического титратора Titroline easy, снабженного магнитной мешалкой и комбинированным электродом.
Результаты исследований и их обсуждение
Изучение ФС на субстанции лекарственных средств показало, что титриметрические методы являются преобладающими при оценке их количественного содержания. Объект нашего исследования – соль органического основания и неорганической кислоты. Для подобных лекарственных средств при количественном определении используется, как правило, метод ацидиметрии в протогенных растворителях. Установлено значение рКа БАС, состав протогенных растворителей, способ индикации. Константу ионизации мономекаина определяли, руководствуясь рекомендациями, изложенными в монографии [1] с использованием титриметрического метода. Изменение pH в процессе титрования регистрировали методом потенциометрии.
Методика: 0,1416 г исследуемого БАС (точная навеска) растворяют в 50 мл воды очищенной (0,01 М концентрация). К полученному раствору десятью порциями, каждая из которых равна одной десятой эквивалента, добавляют титрант – 0,1 М раствор калия гидроксида, перемешивают и после каждого добавления, как только установится равновесие, фиксируют значение pH. Среднее значение рКа, определенное для образцов трех серий БАС, составило 7,52 ± 0,17, что свидетельствует о его достаточно слабых основных свойствах. Исходя из этого, целесообразной является разработка методики его количественного определения на основе метода неводной ацидиметрии.
Анализ методик количественного определения солей органических азотсодержащих оснований, приведенных в действующих фармакопейных статьях, показал, что наиболее распространенными протогенными растворителями являются уксусная кислота ледяная, уксусный ангидрид, муравьиная кислота, а также их смеси [6]. Муравьиная кислота нами была исключена из исследования ввиду ее восстановительных свойств и возможности взаимодействия с нитрат-ионом. Апробированные при разработке методики растворители приведены в табл. 1.
Критериями выбора оптимального растворителя для количественного определения служили следующие: наличие четкого скачка на кривой титрования; количественное содержание БАС, рассчитанное по результатам титрования. Навеску брали с таким расчетом, чтобы на титрование расходовалось 5,0–5,5 мл титранта – 0,1 М раствора хлорной кислоты.
Методика исследования. Около 0,15 г (точная навеска) мономекаина растворяли в каждом из указанных в табл. 1 протогенных растворителях (или их смесях) и титровали 0,1 М раствором хлорной кислоты, добавляя титрант по 0,1 мл, после перемешивания раствора фиксировали значение Е, mv. Вблизи точки эквивалентности добавление титранта проводили по 0,01 мл.
Воспроизводимость полученных результатов, наличие четкого скачка на кривой титрования позволили нам рекомендовать для дальнейших исследований в качестве протогенного растворителя смесь уксусная кислота ледяная – уксусный ангидрид 5:10 (рисунок).
Для визуального установления точки эквивалентности при титровании применяли следующие индикаторы: кристаллический фиолетовый, тропеолин 00, нейтральный красный, малахитовый зеленый. Установлено, что ни один из исследованных индикаторов не дает при титровании мономекаина перехода окраски, обозначенного в контрольном опыте, результаты невоспроизводимы, что, очевидно, связано с влиянием нитрат-иона. Можно предположить, что азотная кислота реагирует с индикаторами, что приводит к изменению их структуры, имеющую иную окраску, чем предусмотренную результатами контрольного опыта. Исходя из этого, нами рекомендован потенциометрический способ фиксирования точки эквивалентности.
Таблица 1
Растворители, использованные для титрования мономекаина
|
№ п/п |
Протогенные растворители |
Соотношение растворителей, мл |
|
1 |
Уксусная кислота ледяная / Уксусный ангидрид |
5:10 |
|
2 |
Уксусная кислота ледяная / Уксусный ангидрид |
10:10 |
|
3 |
Уксусная кислота ледяная / Уксусный ангидрид |
10:5 |
|
4 |
Уксусная кислота ледяная |
10 |
|
5 |
Уксусный ангидрид |
10 |
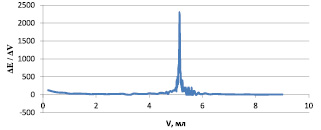
Дифференциальная кривая титрования мономекаина в смеси растворителей: уксусная кислота ледяная – уксусный ангидрид 5:10
Методика ацидиметрического титрования мономекаина в неводной среде:
Около 0,15 г (точная навеска) исследуемого вещества, предварительно высушенного до постоянной массы, растворяют в 5 мл уксусной кислоты ледяной, прибавляют 10 мл уксусного ангидрида и титруют 0,1 М раствором хлорной кислоты. Точку эквивалентности определяют потенциометрически. 1 мл 0,1 М раствора хлорной кислоты соответствует 28,33 мг С13H20ON2·HNO3.
Для подтверждения пригодности разработанной методики проведена ее валидация по показателям: линейность, сходимость и правильность результатов.
Для установления линейной зависимости осуществляли статистическую обработку выборки, полученной в результате количественного определения навесок на семи уровнях концентрации (в диапазоне 70–130 % от количества исследуемого вещества, принятого за 100 % – 0,15 г). Полученное при обработке результатов уравнение линейной регрессии имеет вид: у = 34,63x – 0,1139, а значение коэффициента корреляции (r) составило 0,9998, что подтверждает пригодность методики для количественного определения мономекаина в широком диапазоне концентраций.
Сходимость (повторяемость) результатов оценивали путем статистической обработки данных, полученных в ходе количественного определения исследуемого БАС на трех уровнях концентрации в пределах рекомендуемой аналитической области методики (80, 100, 120 % от количества вещества, принятого за 100 % – 0,15 г.) (табл. 2).
Как следует из представленных данных, относительное стандартное отклонение (RSD) не превышает 0,042 %, что свидетельствует об удовлетворительной сходимости результатов титрования на всех изученных уровнях содержания анализируемого вещества в пробе и соответствии методики критерию приемлемости (не более 1,3 %) при содержании вещества в исследуемом объекте, близком к 100 %.
Правильность методики оценивали по результатам титрования образцов трех серий мономекаина путем статистической обработки результатов титрования (табл. 3). Как следует из полученных результатов, найденное содержание мономекаина близко к 100 %, а величины относительных погрешностей невелики.
Таблица 2
Оценка сходимости результатов титрования (серия БАС 250712)
|
Уровень содержания БАС, % |
Метрологические характеристики (Р = 95 %, f = 6) |
|||
|
Rср, % |
SD |
RSD, % |
∆R |
|
|
80 |
99,99 |
0,042 |
0,042 |
0,08 |
|
100 |
99,87 |
0,035 |
0,035 |
0,07 |
|
120 |
99,97 |
0,039 |
0,039 |
0,06 |
Таблица 3
Результаты количественного определения субстанции мономекаина (Р = 95 %; f = 6)
|
Серия исследуемого БАС |
|
S |
|
|
|
|
080813 |
99,88 |
0,090 |
0,034 |
0,084 |
0,084 |
|
080114 |
99,88 |
0,075 |
0,029 |
0,070 |
0,070 |
|
170314 |
99,87 |
0,083 |
0,031 |
0,070 |
0,077 |



 , %
, %
Заключение
На стадии доклинических испытаний БАС как потенциального лекарственного средства необходима разработка для него стандарта качества в соответствии с установленными требованиями [6]. Одним из основных его разделов является методика количественного определения. В результате проведенных исследований установлены оптимальные условия определения количественного содержания изучаемого БАС методом неводной ацидиметрии, включающие количество БАС, расходуемое на определение, состав протогенного растворителя, способ индикации. Межлабораторное испытание воспроизводимости методики, проведенное в Испытательной лаборатории государственного бюджетного учреждения здравоохранения Свердловской области «Центр контроля качества и сертификации лекарственных средств» (г. Екатеринбург), показало положительные результаты, а ее валидационная оценка по показателям линейность, сходимость и правильность свидетельствует о том, что она соответствует критериям приемлемости для аналитической цели. Исходя из этого, методика нами включена в проект ФС для количественного определения мономекаина и используется для контроля качества лабораторных образцов мономекаина, предназначенных для углубленных фармакологических испытаний и разработки лекарственных форм.
Рецензенты:
Ярыгина Т.И., д.фарм.н., профессор кафедры фармацевтической химии факультета очного обучения, ГБОУ ВПО «Пермская государственная фармацевтическая академия» Министерства здравоохранения Российской Федерации, г. Пермь;
Гейн В.Л., д.х.н., профессор, заведующий кафедрой общей и органической химии, ГБОУ ВПО «Пермская государственная фармацевтическая академия» Министерства здравоохранения Российской Федерации, г. Пермь.
Работа поступила в редакцию 10.04.2015.
Библиографическая ссылка
Чекрышкина Л.А., Бабикова Е.А., Слепова Н.В. КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ МОНОМЕКАИНА МЕТОДОМ НЕВОДНОЙ АЦИДИМЕТРИИ // Фундаментальные исследования. 2015. № 2-15. С. 3333-3337;URL: https://fundamental-research.ru/ru/article/view?id=37780 (дата обращения: 10.07.2026).