



Известно, что хлорид цинка находит широкое применение для улавливания химически связанной серы, а также как один из методов обессеривания нефтяных дестиллятов и малосернистых топлив [2]. При этом образуется сульфид цинка. До настоящего времени механизм протекающих процессов был изучен недостаточно, между тем точное представление о путях возможного взаимодействия компонентов очень важно при разработке технологий неорганических сульфидов в присутствии активатора хлорида цинка.
Исследования серосодержащих систем с использованием современных методов физико-химического анализа и их интерпретация зачастую затруднительны, поэтому в работе предпринята попытка квантово-химического моделирования процессов взаимодействия хлорида цинка с серой.
Цель работы: подбор метода, базиса и исследование взаимодействия компонентов в системе «сера‒хлорид цинка» с использованием квантово-химических расчетов.
Важной предпосылкой успешного квантово-химического исследования является корректность выбора метода расчёта, прежде всего способа учёта электронной корреляции и применяемого базиса. Основными критериями выбора той или иной программы является правильность передачи характера изменения энергии при разрыве связи в различных молекулах и точность передачи геометрической структуры. Расчеты выполнены прикладными программами Gaussian [4] с использованием гибридного метода функционала плотности (B3LYP) и базиса 6-31G* и полуэмпирического метода (PM3), а также программой Priroda [6] с использованием теории функционала плотности (DFT), неэмпирического обменно-кореляционного функционала PBE, в базисном наборе basis4.in, включающего релятивистские поправки, set = L11. При исследовании наиболее выгодного механизма взаимодействия серы с хлоридом цинка была проведена оценка структуры переходных состояний (ПС) и барьеров реакций. Применялся также алгоритм релаксированного сканирования (Scan) геометрических параметров для приближения к предполагаемым переходным состояниям. Чтобы подтвердить принадлежность ПС исследуемому процессу выполнялись спуски по пути реакции к реагентам и продуктам. Результаты и сравнение их с известными литературными данными приведены в табл. 1.
Таблица 1
Энергии диссоциации (D) и длины связей (r) некоторых соединений
|
Соединение |
Литературные данные [1] |
Расчётные данные в Gaussian базис PM3 |
Расчётные данные в Gaussian базис B3LYP/6-31G(d,p) |
Расчётные данные в Gaussian базис B3LYP/6-311++G(df) |
Расчётные данные в Priroda |
|||||
|
r, пм |
D, кДж/ моль |
r, пм |
D, кДж/моль |
r, пм |
D, кДж/моль |
r, пм |
D, кДж/моль |
r, пм |
D, кДж/моль |
|
|
S2 |
188,9 |
422,14 ± 6,3 |
188,9 |
435,85 |
188,9 |
397,2 |
189 |
397,4 |
193 |
434,5 |
|
SH |
134,1 |
340,6 ± 12 |
130,2 |
350,2 |
131 |
346,3 |
131 |
336,7 |
136 |
348,2 |
|
SO |
148,9 |
516,2 ± 0,13 |
148,1 |
619,59 |
148,1 |
754,9 |
148 |
636,8 |
153 |
582,5 |
|
SiS |
192,9 |
619 ± 12,6 |
192,9 |
810,17 |
192,9 |
681,3 |
193 |
677,1 |
197 |
634,3 |
|
SiO |
150,9 |
803,24 ± 21,3 |
150,9 |
970,92 |
150,9 |
734 |
151 |
739,4 |
155 |
875,5 |
|
ZnO |
191,0 |
271,96 ± 41,8 |
191,0 |
283,55 |
191 |
306,4 |
191 |
230,6 |
191 |
290,3 |
|
ZnCl |
224,0 |
221,75 ± 8,37 |
224,0 |
259,3 |
224 |
233,5 |
224 |
190,2 |
218 |
204,7 |
|
ZnS |
198,0 |
200,83 ± 12,5 |
198,9 |
189,76 |
198,9 |
169,5 |
199 |
114,7 |
107 |
187 |
Для сравнения результатов расчета использовали средние арифметические значения затраченного времени на расчёты (τ) и отклонения полученных энергий диссоциации (D) от литературных данных по формуле (1, 2):
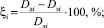 (1)
(1)
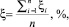 (2)
(2)
где ξi – i-е отклонение экспериментальных данных от литературных, %; ξ – среднее арифметическое отклонение экспериментальных данных, полученных той или иной программой, от литературных данных, %; Dлi – i-е литературное значение энергии диссоциации соединения, кДж/моль; Dэi – i-е экспериментальное значение энергии диссоциации соединения, полученное той или иной программой, кДж/моль; n – количество используемых соединений.
Полученные результаты приведены в табл. 2.
Таблица 2
Сравнение программ по критериям времени и точности
|
Программа, базис |
τ, мин |
ξ, % |
|
Gaussian 09W PM3 |
5,67 |
23,44 |
|
Gaussian 09W B3LYP/6-31G(d,p) |
7,5 |
12,65 |
|
Gaussian 09W B3LYP/6-311++G(df) |
10,4 |
17,3 |
|
Priroda |
9,57 |
8 |
Таким образом, погрешность экспериментальных данных составляет 1–6 %, что не превышает погрешности известных литературных данных.
Поскольку наиболее точными и экономичными по временным затратам являются расчеты с применением программы Priroda, нами для дальнейших исследований использовался этот метод.
Исследование двойной системы
Для оценки наиболее выгодного механизма взаимодействия серы с хлоридом цинка была проведена оценка структуры переходных состояний и барьеров реакций. Все расчеты проводились для синглетного состояния. Выполнен анализ присоединения одноатомной, двух-, четырех- и шестиатомной серы к активирующей добавке. Присоединение одноатомной серы к хлориду цинка происходит безактивационно, с большим выделением тепла –219,16 кДж/ моль.
Реакция присоединения двухатомной серы к хлориду цинка (рис. 1) является экзотермической. Энергия активации составляет 27,24 кДж/моль, т.е. взаимодействие протекает легче. В переходном состоянии происходит поворот ближайшего атома хлора к атому серы. В результате спуска образуется связь S–Cl (210,9 пм), а связь Zn–S укорачивается до 218,5 пм. Прочность сформировавшейся связи Zn–S составляет 219,3 кДж/моль.
Схожий механизм можно увидеть и при присоединении четырехатомной серной молекулы к хлориду цинка (рис. 2). Молекула серы закрепляется к хлориду цинка в виде разомкнутого цикла, где цинк повышает свою координацию до четырех. Затем в переходном состоянии серное кольцо раскрывается с приближением конечной серы к атому хлора. В продукте реакции связи S–Cl и Zn–S укорачивается и составляет 213 и 222,5 пм соответственно. Прочность сформировавшейся связи Zn–S составляет 247 кДж/моль. Энергия активации данного процесса близка к энергии присоединения двухатомной серы и составляет 26,82 кДж/моль.
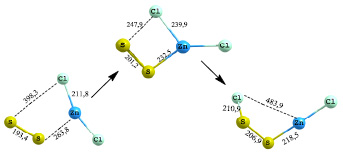
Рис. 1. Схема присоединения двухатомной серы к хлориду цинка
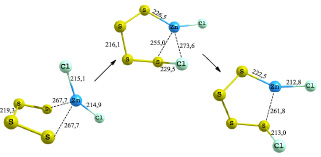
Рис. 2. Схема присоединения четырехатомной серы к хлориду цинка
Известно [5], что наиболее стабильными циклическими формами серы являются молекулы с высокой симметрией: S8 в виде короны и S6 в виде кресла.
В результате оптимизации исходного комплекса (рис. 3) молекула S6 присоединяется к хлориду цинка в виде замкнутого цикла. Более близкое приближение атома цинка к сере, по-видимому, вызвано стерическим препятствием, формируется длинная связь Zn–S – 261,4 пм, а для сульфидов свойственна связь Zn–S, равная 217 пм. Прочность образовавшейся связи равна 29 кДж/моль. В переходном состоянии цикл S6 раскрывается, связь Zn-Cl удлиняется до 229,6 пм и появляется вероятность ее разрыва, связь Zn–S укорачивается до 234,5 пм. В результате спусков получен сложный сульфидный комплекс, где формируются короткие связи Zn–S и S–Cl, которые составляют 223,2 и 210,2 пм соответственно. Прочность образовавшейся связи Zn–S равна 262,7 кДж/моль. Энергия активации данного процесса составляет 93,63 кДж/моль. По сравнению с процессом присоединения хлорида цинка к S2 и S4 представленны в табл. 3 энергетический барьер присоединения активатора к S6 выше. поскольку взаимодействие протекает в две стадии (раскрытие цикла S6 и переход атома хлора к сере). Однако он легко преодолевается в температурных условия синтеза.
Можно предположить, что присоединение восьмиатомной серы будет происходить аналогично присоединению молекулы S6.
Таблица 3
Тепловой эффект и энергия активации присоединения серы S1, S2, S4, S6
|
Процесс |
∆Hреакции, кДж/моль |
Еакт,кДж/моль |
|
Присоединение атомарной серы |
–219,16 |
0 |
|
Присоединение двухатомной серы |
–12,43 |
27,24 |
|
Присоединение четырехатомной серы |
20,84 |
26,82 |
|
Присоединение шестиатомной серы |
55,14 |
93,63 |
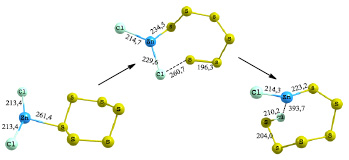
Рис. 3. Схема присоединения шестиатомной серы к хлориду цинка
Результаты квантово-химических расчетов были подтверждены результатами исследований свойств физико-химического анализа (ИК-спектроскопия, исследование реологических свойств, рентгенофазовый анализ [7]).
Таким образом, предложена квантово-химическая модель процесса взаимодействия хлорида цинка с серой, заключающаяся в дестабилизации серного компонента, раскрытия молекул с образованием реакционно-активных радикалов и последующего присоединения к хлориду цинка с образованием устойчивых молекул сульфида цинка.
Основные результаты и выводы
1. Исследовано взаимодействие хлорида цинка к молекулам серы: S, S2, S4, S6.
2. В результате взаимодействия активатора хлорида цинка с серой образуются стабильные сульфидные компоненты с энергией связи Zn–S порядка 219–263 кДж/моль, что указывает на их высокую термическую стабильность полученных сульфидов цинка.
3. Квантово-химические расчеты показали, что активатор хлорида цинка способствует снижению энергии разрыва серного кольца S6. Таким образом, можно утверждать, что активатор способствует дестабилизации циклов, активирует их разрыв и образование радикалов.
Рецензенты:
Сайфуллин Р.С., д.т.н., профессор кафед_ры «Технологии неорганических веществ и материалов», ФГБОУ ВПО «КНИТУ», г. Казань;
Корнилов А.В., д.т.н., профессор, зав. отделом технологических испытаний, ФГУП ЦНИИ «Геолнеруд», г. Казань.
Работа поступила в редакцию 30.04.2013.
Библиографическая ссылка
Сабахова Г.И КВАНТОВО-ХИМИЧЕСКОЕ МОДЕЛИРОВАНИЕ ПРОЦЕССА ВЗАИМОДЕЙСТВИЯ ХЛОРИДА ЦИНКА С СЕРОЙ // Фундаментальные исследования. 2013. № 6-5. С. 1137-1140;URL: https://fundamental-research.ru/ru/article/view?id=31702 (дата обращения: 14.07.2026).