



Для хелатных комплексов редкоземельных элементов (РЗЭ) характерно наличие интенсивной люминесценции [1, 2]. Исследования явления люминесценции актуальны с фундаментальной (изучение механизма преобразования световой энергии) и с прикладной точек зрения (разработка многофункциональных светотрансформирующих материалов нового поколения). Ранее авторами были проведены экспериментальные исследования электронных переходов и квантово-химическое моделирование электронной структуры, возбужденных состояний и спектров поглощения хелата лантана (III) состава La(NO3)3(ГМФА)3 [4, 5] и фосфаалкенов [3, 9, 11], являющихся перспективными лигандами в химии комплексных соединений РЗЭ.
Целью настоящей работы является теоретическое физико-химическое исследование структуры, электронного строения и спектрального поведения хелатов иттрия состава Y(NO3)3(ГМФА)3 (I) и Y(БТФА)2NO3(ТФФО)2 (II) (БТФА – C6H5COCHCOCF3, бензоилтрифторацетонат-анион, ТФФО – OP(C6H5)3, трифенилфосфиноксид).
Материалы и методы исследования
С помощью программы GAMESS-US [8] методами функционала плотности DFT и TDDFT с гибридным обменно-корреляционным функционалом PBE0 [6], Штутгартским псевдопотенциалом и базисом ECP-MWB [10] в вакуумном приближении выполнены квантово-химические расчеты электронного строения и физико-химических свойств хелатных комплексов I и II в основном и возбужденных состояниях.
Результаты исследования и их обсуждение
Согласно проведенным расчетам методом DFT, модельная геометрическая структура комплексов I, II согласуется с экспериментальными данными для изоструктурных лантанидных комплексов аналогичного состава [1, 2]. Оценка параметров электронного строения кристаллической фазы проведена на основе квантово-химических расчетов соединений Y(NO3)3ГМФА3 и Y(БТФА)2NO3(ТФФО)2. Расчетные порядки связей комплекса II при переходе от экспериментальной к оптимальной геометрии изменяются мало, но в комплексе I порядки связей Y-O с нейтральными лигандами ГМФА уменьшаются, что подтверждается ростом длин этих связей.
Квантово-химическое моделирование показало, что данные молекулярные системы являются сильно полярными, их дипольный момент равен 9,6 и 10,5 Д соответственно. Установлено, что распределение электронной плотности комплексов I и II при переходе от оптимальной к экспериментальной геометрии изменяется незначительно, заряд РЗ атома уменьшается на 0,40е и 0,14е соответственно. При этом изменении геометрии дипольный момент молекул увеличивается на 5,9 и 5,0 Д соответственно, что свидетельствует о существенном росте поляризации молекулы при переходе в кристаллическую фазу.
Состав граничных молекулярных орбиталей (МО) (рис. 1) и энергетические щели между ними позволили определить параметры электронного строения молекулярных систем I, II.
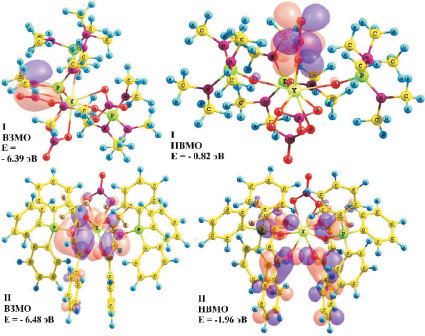
Рис. 1. Энергии и состав граничных МО комплексов I и II
Согласно расчетным данным, верхняя занятая МО (ВЗМО) комплекса I с энергией – 6,39 эВ является nN-МО, характеризующей неподеленную электронную пару (НЭП) атома азота лиганда ГМФА, а ВЗМО комплекса II с энергией –6,48 эВ является π-МО, характеризующей π–систему –C(O)CHC(O)– хелатного кольца бензоилтрифторацетоната. Нижняя вакантная МО (НВМО) комплекса I с энергией –0,82 эВ является π*-МО, характеризующей π–систему группы NO3, а НВМО комплекса II с энергией –1,96 эВ является π*-МО, характеризующей π–системы бензоилтрифторацетонатов. Разности энергий ВЗМО и НВМО комплексов I и II составляют 5,57 и 4,52 эВ соответственно. Из состава граничных МО следует, что электронное возбуждение данных молекулярных систем будет определяться в основном свойствами лигандов, т.е. в электронных спектрах поглощения и люминесценции вероятно появление интенсивных пиков, обусловленных возбуждением электронов лигандов.
Методом TDDFT/PBE0/ECP-MWB проведен расчет 20 возбужденных синглетных и триплетных состояний хелатных комплексов I и II, смоделированы их УФ-спектры поглощения (рис. 2). Сопоставление результатов расчетов с экспериментальными спектрами поглощения соединений I и II показало следующее.
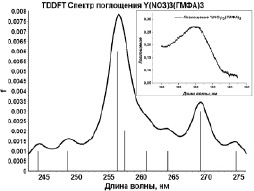
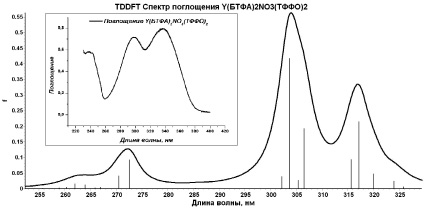
Рис. 2. Модельные спектры поглощения комплексов I и II (На вкладках – экспериментальные спектры поглощения [5])
Согласно квантово-химическим расчетам электронных синглет–синглетных переходов комплекса Y(NO3)3(ГМФА)3, возбуждение молекулы в синглетном состоянии более чем на 80 % обусловлено переходами валентных электронов, соответствующих НЭП атомов азота и СН-связям нейтральных лигандов ГМФА, на вакантные молекулярные уровни, соответствующие несвязывающей π*-МО π–систем групп NO3. Таким образом, наиболее вероятные центры электронного возбуждения при переходе комплекса I в возбужденные состояния локализованы на лигандах. На основании этого можно предположить, что широкая полоса с максимальной интенсивностью (λпогл = 300 нм) экспериментального спектра поглощения комплекса I обусловлена электронными переходами в лигандах.
По данным расчетов э соединения Y(БТФА)2NO3(ТФФО)2, возбуждение молекулярной системы в синглетном состоянии более чем на 90 % обусловлено переходами валентных электронов, соответствующих π–системам C(O)CHC(O) хелатного кольца и фенильного кольца бензоилтрифторацетоната, на вакантные молекулярные уровни, соответствующие несвязывающим π*-МО π–систем этих колец. Очевидно, что наиболее вероятные центры электронного возбуждения при переходе комплекса II в возбужденные синглетное и мультиплетные состояния локализованы именно на лигандах. Можно предположить, что интенсивные полосы (λпогл = 317, 307 и 273 нм) экспериментального спектра поглощения данного хелатного комплекса также обусловлены электронными переходами в лигандах.
Анализ электронных синглет–синглетных переходов изученных молекулярных систем из возбужденного состояния в основное состояние показал, что в соединении I эти процессы в основном обусловлены переходами валентных электронов с вакантных молекулярных уровней, соответствующих несвязывающей π*-МО π-систем групп NO3, на уровни, соответствующие nN- и σСН-МО, отвечающим НЭП атомов азота и СН-связям ГМФА. Полоса флуоресценции с максимальной интенсивностью (λфлуор = 430 нм) экспериментального спектра комплекса I обусловлена этими электронными переходами (рис. 3).
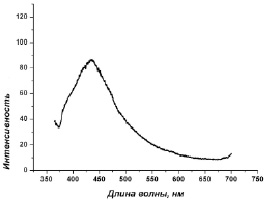
Рис. 3. Спектр люминесценции комплекса I (λвозб = 300 нм) [5]
В соединении II переходы системы из возбужденного в основное состояние связаны с переходами валентных электронов с вакантных молекулярных уровней, соответствующие несвязывающим π*-МО π–систем C(O)CHC(O) хелатного кольца и фенильного кольца бензоилтрифторацетоната, на уровни, соответствующие π–МО этих колец. Таким образом, полосы флуоресценции с максимальной интенсивностью экспериментального спектра люминесценции комплекса II также обусловлены электронными переходами в лигандах.
Выводы
Методами функционала плотности DFT и TDDFT/PBE0/ECP-MWB в вакуумном приближении смоделированы структура и спектральные свойства хелатных комплексов иттрия Y(NO3)3(ГМФА)3 и Y(БТФА)2NO3(ТФФО)2. Выполнены квантово-химические расчеты геометрического и электронного строения хелатов иттрия в основном и возбужденных состояниях. Показано, что изученные молекулярные системы являются полярными, их дипольный момент равен 9,6 и 10,5 Д соответственно. На основе моделирования дана интерпретация особенностей экспериментальных электронных спектров поглощения и люминесценции, сделаны выводы о механизме люминесценции.
Работа проводилась при частичной финансовой поддержке Министерства образования и науки Российской Федерации в рамках государственного задания Дальневосточного федерального университета № 3.2261.2011.
Рецензенты:
Игнатьева Л.Н., д.х.н., заведующая лабораторией фторидных материалов, ФГБУН «Институт химии ДВО РАН», г. Владивосток;
Кавун В.Я., д.х.н., заведующий лабораторией химической радиоспектроскопии, ФГБУН «Институт химии ДВО РАН», г. Владивосток.
Работа поступила в редакцию 07.05.2013.
Библиографическая ссылка
Харченко В.И., Алексейко Л.Н., Мирочник А.Г., Жихарева П.А., Чередниченко А.И. КВАНТОВО-ХИМИЧЕСКОЕ ИССЛЕДОВАНИЕ ЭЛЕКТРОННОГО СТРОЕНИЯ И ВОЗБУЖДЕННЫХ СОСТОЯНИЙ ХЕЛАТНЫХ КОМПЛЕКСОВ ИТТРИЯ // Фундаментальные исследования. 2013. № 6-4. С. 901-905;URL: https://fundamental-research.ru/ru/article/view?id=31659 (дата обращения: 19.07.2026).