



Адренергическая гиперреактивность рассматривается как один из ключевых звеньев в современной концепции генеза различных форм артериальной гипертонии (АГ). В наших исследованиях на модели стресс-индуцированной АГ была выявлена взаимосвязь между высокой кардиоваскулярной стресс-реактивностью у гипертензивных объектов и повышением плотности/чувствительности адренорецепторов, которые вовлекаются в прессорные и хронотропные эффекты стресса. Наиболее важным открытием в этой области явился тот факт, что уже на латентной стадии развития стресс-индуцированной АГ у животных отмечаются изменения в кардиоваскулярной стресс-реактивности, подобные таковым у крыс с устойчивой формой болезни - прессорные эффекты стресса, не изменяясь по интенсивности, существенно пролонгируются во времени [1]. Исследуя причины данного феномена с акцентом на изучении роли адренергических влияний, мы не обнаружили каких-либо изменений в прессорных механизмах активации адренорецепторов на стадии предгипертонии. Однако в этих условиях чувствительность β2-адренорецепторов, обеспечивающих вазорелаксацию, существенно снижается и прогрессирует по мере развития заболевания. Отметим, что нарушения адренозависимой вазорелаксации констатируются и в клинических исследованиях у людей, предрасположенных к АГ по наследственному фактору [7]. За последние годы в научной литературе накоплено большое количество данных, отражающих перспективность использования фармакологического тестирования чувствительности β2-адренорецепторов к их стимуляции в качестве маркера ранних форм гипертонии [5]. В опытах с подсадкой гена с повышенной экспрессией β2-адренорецепторов в сосуды гипертензивным объектам была показана перспективность использования такого воздействия в качестве антигипертензивной терапии [4]. Эти факты делают актуальным изучение механизмов, лежащих в основе нарушения адренозависимой вазорелаксации при развитии АГ.
Поскольку акцент научных исследований многие годы был сконцентрирован на изучении механизмов прессорных эффектов симпатических влияний на сердечно-сосудистую систему, работы, посвященные анализу причин нарушения адренозависимой вазорелаксации у гипертоников носили в основном феноменологический характер.
Что известно в этой области к настоящему времени? В ряде работ была выявлена взаимосвязь между эндотелиальными (в частности, оксидом азота (NO)) и адренергическими механизмами вазорелаксации [3]. С использованием радиологических методов была показана топография β2- адренорецепторов непосредственно на эндотелии [8]. Однако некоторые ученые подвергают сомнению указанную точку зрения, не обнаружив какой-либо связи между NO-ергическими влияниями и деятельностью β2-адренорецепторов [6]. В последние годы сформировалась устойчивая тенденция к признанию важной роли КАТФ-каналов в механизмах адренозависимой вазорелаксации [9]. Таким образом, противоречивость экспериментальных данных затрудняет формирование обобщенной точки зрения о механизмах β2-адренозависимой вазодилатации и особенно их роли в формировании гипертензивного статуса.
Цель исследований
Изучение механизмов β2-адренозависимой вазорелаксации в норме и при развитии артериальной гипертонии.
Материалы и методы
Исследования выполнены на 97 нормотензивных и гипертензивных [2] самцах крыс. Запись сигналов кровяного давления осуществляли на компьютерно-вычислительном комплексе для прямой регистрации артериального давления (ср. АД) у мелких животных (Power Lab/400 ML 401, ID Instruments, 2002, Австралия) с программным обеспечением Chart 4, оснащенном датчиками кровяного давления (MLT0699, Power Lab, ID Instruments). Введение фармакологических препаратов: изопротеренола - β2-адреноагонист (Sigma, 0,5 мкг/кг, 0,05 мкг/кг, 0,005 мкг/кг, iv), глибенкламида - блокатор КАТФ-каналов (Sigma, 2,5 мг/кг, 5 мг/кг, 10 мг/кг, iv), индометацина - блокатор простациклина PGI2 (Sopharma, Bulgaria, 1 мг/кг, 2,5 мг/кг, 5 мг/ кг, iv), NG-nitro-L-arginine-methyl ester - блокатор NO-синтазы (L-NAME, Sigma, 2,5 мг/кг, 5 мг/кг, 10 мг/кг, iv) осуществляли через полиэтиленовый катетер, вживленный в яремную вену под общим нембуталовым наркозом (0,40 мг/кг, ip). Для статистической обработки использовали пакет программ Statistica 5.0. Различия считали достоверными при р<0,05.
Результаты исследования
Введение изопротеренола, стимулирующего β2-адрено-зависимую вазорелаксацию, сопровождалось развитием гипотензивных реакций. При этом уже на начальных стадиях формирования гипертензивного статуса (предгипертензивная группа) отмечалось снижение сосудистой чувствительности к изопротеренолу. По мере развития заболевания наблюдалось прогрессирование указанных нарушений. Так, у животных с устойчивой артериальной гипертонией отмечалось более существенное снижение гипотензивных эффектов изопротеренола по сравнению с контролем, чем в предгипертензивной группе (рис. 1).
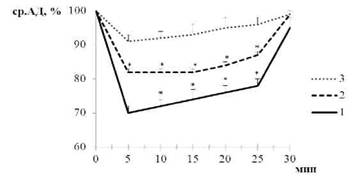
Рис. 1. Изменение среднего артериального давления (ср.АД, %) под влиянием изопротеренола, p<0,05 относительно: * - базальных значений, † - нормотензивной группы
1 - нормотензивная группа; 2 - предгипертензивная группа; 3 - гипертензивная группа
Поскольку основными механизмами вазорелаксации является активация эндотелиальных факторов, включающих NO и PGI2, а также стимуляцию К+-каналов, в данных опытах изучалось влияние блокады этих трех ключевых звеньев вазодилатации на сосудистые эффекты изопротеренола у нормотензивных и гипертензивных животных.
У нормотензивных крыс предварительная блокада NO-синтазы путем введения L-NAME вызывала дозозависимое снижение сосудистой чувствительности к изопротеренолу (рис. 2а). Предварительная блокада КАТФ-каналов глибенкламидом также индуцировала дозозависимое подавление гипотензивных эффектов изопротеренола (рис. 2б).
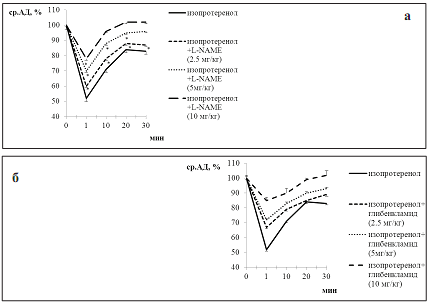
Рис. 2. Изменение среднего артериального давления (ср.АД, %) под влиянием изопротеренола на фоне предварительной блокады NO-синтазы (А) и КАТФ-каналов (Б) у нормотензивных крыс; * - p<0,05 относительно базальных значений
Предварительная блокада PGI2 индометацином не сопровождалась какими-либо изменениями в сосудистых эффектах изопротеренола. Гипотензивные реакции на фоне введения индометацин+изопротеренол были примерно одинаковыми как по амплитуде, так и по длительности по сравнению с группой крыс, получающих только изопротеренол.
Поскольку в ходе эксперимента были выявлены эффекты блокады продукции NO и активности КАТФ-каналов, а не PGI2 на сосудистую чувствительность к изопротеренолу, далее тестировались гипотензивные реакции изопротеренола на фоне одновременного введения L-NAME (10 мг/ кг) и глибенкламида (10 мг/кг). Предварительная блокада синтеза NO и КАТФ-каналов полностью предотвращала способность изопротеренола к вазорелаксации (рис.3).
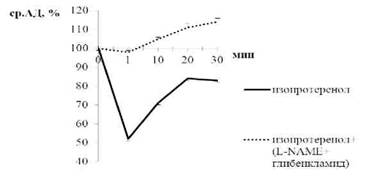
Рис. 3. Изменение среднего артериального давления (ср.АД, %) под влиянием изопротеренола на фоне двойной блокады NO-синтазы и КАТФ-каналов у нормотензивных крыс; * - p<0,05 относительно базальных значений
Таким образом, результаты экспериментов показали, что дилатация сосудов, вызванная активацией бета2-адренорецепторов, осуществляется активацией КАТФ-каналов и синтеза NO, но не PGI2.
В ряде исследований, направленных на изучение взаимосвязи между β2- адренозависимыми и эндотелиальными/неэндотелиальными механизмами вазорелаксации, было обнаружено, во-первых, что введение различных блокаторов синтеза NO частично ингибирует гипотензивные эффекты препаратов, стимулирующих активность β2- адренорецепторов [3], а во-вторых, в опытах in vitro на коронарных артериях кошек было обнаружено, что изопротеренол усиливает эффекты ацетилхолина за счет повышения активности КАТФ-каналов [9]. Эти факты органично согласуется с результатами, полученными в ходе данного этапа экспериментов.
На основе полученных результатов, свидетельствующих о вовлечении КАТФ-каналов и NO в механизмы бета2-адренозависимой вазорелаксации, в следующей серии опытов изучалась активность этих факторов у крыс на разных этапах развития АГ.
Результаты исследования показали, что уже на начальных этапах формирования АГ отмечалось снижение сосудистой чувствительности к блокаде КАТФ-каналов. Действительно, в предгипертензивной группе амплитуда снижении ср.АД глибенкламидом была ниже в 2,2 раза (p<0,05), чем у нормотензивных крыс (рис. 4а). Однако по длительности гипотензивных эффектов глибенкламида не были обнаружены различия между экспериментальными группами.
Прогрессирование АГ сопровождалось усугублением нарушений в механизмах вазорелаксации, связанных с активацией КАТФ- каналов. Так, амплитуда снижения ср.АД в ответ на введение глибенкламида у гипертензивных крыс была ниже в 3,8 раза (p<0,05) по сравнению с нормотензивными животными (рис. 4а). При этом длительность гипотензивных эффектов глибенкламида у первых была существенно снижена, чем у вторых.
Сосудистая чувствительность к введению L-NAME (рис. 4а,б), равно как и содержание NO в крови, не различались между предгипертензивной и нормотензивной группами (0,42 мкг/мл против 0,38 мкл/ мл - для NO в крови), но существенно снижались у гипертензивных животных (0,42 мкл/мл против 0,22 мкл/мл, p<0,05 - для NO в крови).
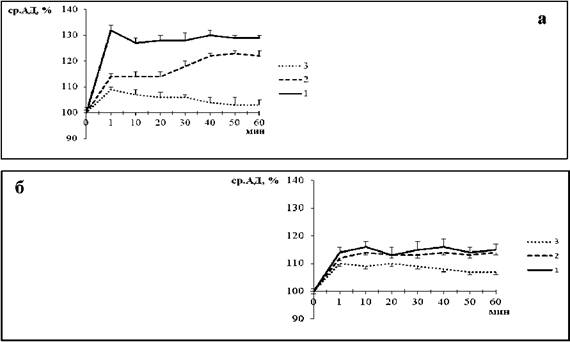
Рис. 4. Изменение среднего артериального давления (ср.АД, %) под влиянием блокады КАТФ-каналов (А) и NO-синтазы (Б) в 1 - нормотензивной, 2 - предгипертензивной, 3 - гипертензивной группах; * - p<0,05 относительно базальных значений, † - нормотензивной группы
Таким образом, механизмы нарушения бета2-адренозависимой вазорелаксации различны на этапах формирования и прогрессирования АГ. Латентная форма АГ характеризуется снижением активности КАТФ-каналов и отсутствием каких-либо изменений в NO-зависимых процессах вазодилатации. Прогрессирование АГ сопровождается усугублением первичных нарушений в деятельности КАТФ- каналов и сопутствующим снижением в активности системы генерации NO.
Список литературы
- Семячкина-Глушковская О.В., Анищенко Т.Г., Бердникова В.А., Кузнецова А.С., Кузнецова Я.В., Семячкин-Глушковский И.А. Кардиоваскулярная стресс-реактивность как индикатор развития гипертонии // Известия Самарского научного центра Российской академии наук. - 2009. - Т. 11, - № 1 (5). - С. 1027-1030.
- Патент РФ № 68280/33, 27.11.2007.
- Dawes M., Chowienczyk P.J., Ritter J.M. Effects of inhibition of the L-arginine/nitric oxide pathway on vasodilation caused by β-adrenergic agonists in human forearm // Circulation. - 1997. - Vol. 95. - P. 2293- 2297.
- Iaccarino G. ß2 -Adrenergic receptor gene delivery to the endothelium corrects impaired adrenergic vasorelaxation in hypertension / G. Iaccarino, E. Cipolletta, A. Fiorillo, et al. // Circulation. - 2002. - Vol. 106. - P. 349.
- Masuo K. ß2- and ß3-adrenergic receptor polymorphisms are related to the onset of weight gain and blood pressure elevation over 5 years / K. Masuo, T. Katsuya, Y. Fu et al. // Circulation. - 2005. - Vol. 111. - P. 3429-3434.
- Moncada S., Rees D.D., Schulz R.M. Palmer Development and mechanism of a specific supersensitivity to nitrovasodilators after inhibition of vascular nitric oxide synthesis in vivo // Proc. Natl. Acad. Sci. USA. - 1991. - Vol. 88. - P. 2166-2170.
- Schlaich M. Beta2-adrenoreceptor-mediated, nitric-oxide-dependent vasodilatation is abnormal in early hypertension: restoration by L-arginine / Ahlers B., Parnell M. // J. Hyper. - 2004, 22(10): 1917-1925.
- Summers R.J., Molenaar R.J., Stephenson J.A., Jones C.R. Autoradiographic localization of receptors in the mammalian cardiovascular system // Clin. Exp. Pharmacol. Physiol. - 1987. - Vol. 14. - P. 437-447.
- Wang Y.G., Lipsius S.L. Beta-adrenergic stimulation induces acetylcholine to activate ATP-sensitive K+ current in cat arterial myocytes // Circ. Res. - 1995. - Vol. 77(3). - P. 565-574.
Библиографическая ссылка
О.В. Семячкина-Глушковская, Т.Г. Анищенко. В.А. Бердникова, Я.В. Кузнецова, А.С. Кузнецова, И.А. Семячкин-Глушковский, С.В. Капралов МЕХАНИЗМЫ НАРУШЕНИЯ АДРЕНОЗАВИСИМОЙ ВАЗОРЕЛАКСАЦИИ ПРИ РАЗВИТИИ АРТЕРИАЛЬНОЙ ГИПЕРТОНИИ // Фундаментальные исследования. 2010. № 2. С. 123-129;URL: https://fundamental-research.ru/ru/article/view?id=1657 (дата обращения: 26.06.2026).